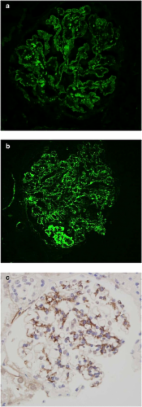
Hemolytic uremic syndrome (HUS) is defined by the triad of mechanical hemolytic anemia, thrombocytopenia and renal impairment. Atypical HUS (aHUS) defines non Shiga-toxin-HUS and even if some authors include secondary aHUS due to Streptococcus pneumoniae or other causes, aHUS designates a primary disease due to a disorder in complement alternative pathway regulation. Atypical HUS represents 5 -10% of HUS in children, but the majority of HUS in adults. The incidence of complement-aHUS is not known precisely. However, more than 1000 aHUS patients investigated for complement abnormalities have been reported. Onset is from the neonatal period to the adult age. Most patients present with hemolytic anemia, thrombocytopenia and renal failure and 20% have extra renal manifestations. Two to 10% die and one third progress to end-stage renal failure at first episode. Half of patients have relapses. Mutations in the genes encoding complement regulatory proteins factor H, membrane cofactor protein (MCP), factor I or thrombomodulin have been demonstrated in 20-30%, 5-15%, 4-10% and 3-5% of patients respectively, and mutations in the genes of C3 convertase proteins, C3 and factor B, in 2-10% and 1-4%. In addition, 6-10% of patients have anti-factor H antibodies. Diagnosis of aHUS relies on 1) No associated disease 2) No criteria for Shigatoxin-HUS (stool culture and PCR for Shiga-toxins; serology for anti-lipopolysaccharides antibodies) 3) No criteria for thrombotic thrombocytopenic purpura (serum ADAMTS 13 activity > 10%). Investigation of the complement system is required (C3, C4, factor H and factor I plasma concentration, MCP expression on leukocytes and anti-factor H antibodies; genetic screening to identify risk factors). The disease is familial in approximately 20% of pedigrees, with an autosomal recessive or dominant mode of transmission. As penetrance of the disease is 50%, genetic counseling is difficult. Plasmatherapy has been first line treatment until presently, without unquestionable demonstration of efficiency. There is a high risk of post-transplant recurrence, except in MCP-HUS. Case reports and two phase II trials show an impressive efficacy of the complement C5 blocker eculizumab, suggesting it will be the next standard of care. Except for patients treated by intensive plasmatherapy or eculizumab, the worst prognosis is in factor H-HUS, as mortality can reach 20% and 50% of survivors do not recover renal function. Half of factor I-HUS progress to end-stage renal failure. Conversely, most patients with MCP-HUS have preserved renal function. Anti-factor H antibodies-HUS has favourable outcome if treated early.