- Record: found
- Abstract: found
- Article: found
BioBin: a bioinformatics tool for automating the binning of rare variants using publicly available biological knowledge
Read this article at
Abstract
Background
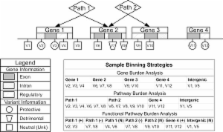
With the recent decreasing cost of genome sequence data, there has been increasing interest in rare variants and methods to detect their association to disease. We developed BioBin, a flexible collapsing method inspired by biological knowledge that can be used to automate the binning of low frequency variants for association testing. We also built the Library of Knowledge Integration (LOKI), a repository of data assembled from public databases, which contains resources such as: dbSNP and gene Entrez database information from the National Center for Biotechnology (NCBI), pathway information from Gene Ontology (GO), Protein families database (Pfam), Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, NetPath - signal transduction pathways, Open Regulatory Annotation Database (ORegAnno), Biological General Repository for Interaction Datasets (BioGrid), Pharmacogenomics Knowledge Base (PharmGKB), Molecular INTeraction database (MINT), and evolutionary conserved regions (ECRs) from UCSC Genome Browser. The novelty of BioBin is access to comprehensive knowledge-guided multi-level binning. For example, bin boundaries can be formed using genomic locations from: functional regions, evolutionary conserved regions, genes, and/or pathways.
Methods
We tested BioBin using simulated data and 1000 Genomes Project low coverage data to test our method with simulated causative variants and a pairwise comparison of rare variant (MAF < 0.03) burden differences between Yoruba individuals (YRI) and individuals of European descent (CEU). Lastly, we analyzed the NHLBI GO Exome Sequencing Project Kabuki dataset, a congenital disorder affecting multiple organs and often intellectual disability, contrasted with Complete Genomics data as controls.
Results
The results from our simulation studies indicate type I error rate is controlled, however, power falls quickly for small sample sizes using variants with modest effect sizes. Using BioBin, we were able to find simulated variants in genes with less than 20 loci, but found the sensitivity to be much less in large bins. We also highlighted the scale of population stratification between two 1000 Genomes Project data, CEU and YRI populations. Lastly, we were able to apply BioBin to natural biological data from dbGaP and identify an interesting candidate gene for further study.
Related collections
Most cited references16

- Record: found
- Abstract: found
- Article: not found
An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people.
- Record: found
- Abstract: found
- Article: not found