- Record: found
- Abstract: found
- Article: not found
The Telomeric Protein TRF2 Binds the ATM Kinase and Can Inhibit the ATM-Dependent DNA Damage Response
Read this article at

Abstract
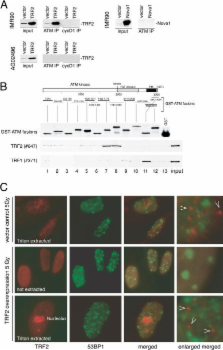
The telomeric protein TRF2 is required to prevent mammalian telomeres from activating DNA damage checkpoints. Here we show that overexpression of TRF2 affects the response of the ATM kinase to DNA damage. Overexpression of TRF2 abrogated the cell cycle arrest after ionizing radiation and diminished several other readouts of the DNA damage response, including phosphorylation of Nbs1, induction of p53, and upregulation of p53 targets. TRF2 inhibited autophosphorylation of ATM on S1981, an early step in the activation of this kinase. A region of ATM containing S1981 was found to directly interact with TRF2 in vitro, and ATM immunoprecipitates contained TRF2. We propose that TRF2 has the ability to inhibit ATM activation at telomeres. Because TRF2 is abundant at chromosome ends but not elsewhere in the nucleus, this mechanism of checkpoint control could specifically block a DNA damage response at telomeres without affecting the surveillance of chromosome internal damage.
Abstract
The human telomere-associated protein TRF2 inhibits ATM kinase, suggesting a mechanism by which the telomeric protein complex prevents the activation of this DNA damage response transducer
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
ATM and related protein kinases: safeguarding genome integrity.
- Record: found
- Abstract: found
- Article: not found