- Record: found
- Abstract: found
- Article: found
IEDB-AR: immune epitope database—analysis resource in 2019

Read this article at
Abstract
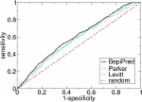
The Immune Epitope Database Analysis Resource (IEDB-AR, http://tools.iedb.org/) is a companion website to the IEDB that provides computational tools focused on the prediction and analysis of B and T cell epitopes. All of the tools are freely available through the public website and many are also available through a REST API and/or a downloadable command-line tool. A virtual machine image of the entire site is also freely available for non-commercial use and contains most of the tools on the public site. Here, we describe the tools and functionalities that are available in the IEDB-AR, focusing on the 10 new tools that have been added since the last report in the 2012 NAR webserver edition. In addition, many of the tools that were already hosted on the site in 2012 have received updates to newest versions, including NetMHC, NetMHCpan, BepiPred and DiscoTope. Overall, this IEDB-AR update provides a substantial set of updated and novel features for epitope prediction and analysis.
Related collections
Most cited references17
- Record: found
- Abstract: found
- Article: not found
NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data.
- Record: found
- Abstract: found
- Article: not found
Gapped sequence alignment using artificial neural networks: application to the MHC class I system.
- Record: found
- Abstract: found
- Article: found