
INTRODUCTION. ASYMMETRIC SYNTHESIS: THE QUERY AND THE OFFER
The observation of symmetry and asymmetry in bodies and, in general, in the whole universe is an ancient matter [1]. In the eighteenth century, Kant wrote that our hands can be thought as mirror images which are not superimposable. The same feature is observed in molecules having a specific property, chirality. The word, first introduced in 1904, derives from Greek ‘χεῖρ’ (‘kheir’, meaning ‘hand’). The discovery of chirality happened in 1815, with the observation by Jean Baptiste Biot that some molecules could rotate the polarised light plane [2].
The occurrence of only one enantiomer of a molecule in nature is connected to the evidence that enantiomers have different biological activity and, in general, they have a different behaviour in a chiral environment [3]. Thus enzymes, receptors and chiral molecules occurring in biochemical transformations, interact in a different way with the left-handed or the right-handed enantiomer of a chemical or drug. For instance, some different fragrances are isomers of the same molecule: (S)-(−)-limonene (1) smells like turpentine, whereas (R)-(+)-limonene (2) smells like lemon (Figure 1a). One can distinguish the two fragrances thanks to our nasal receptors, made of chiral molecules able to recognise the difference between the two enantiomers.
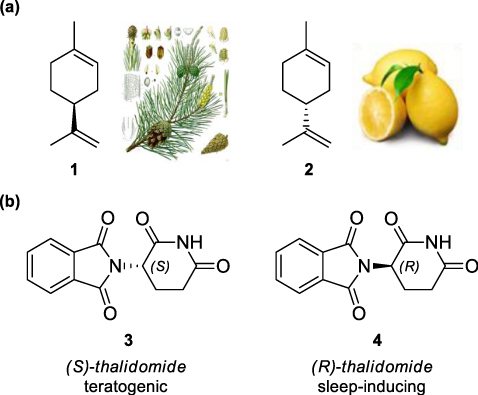
Other examples are molecules used by insects as sexual attractors, pheromones, as well as most of commercialised drugs. Biochemical processes are strongly affected by stereochemical differences because of the chirality of the involved molecules. A tragic example is thalidomide, a sedative drug which was broadly prescribed to pregnant women since it did not cause dependence (Figure 1b): unfortunately, one of the two enantiomers was responsible for foetus deformities as teratogen agent [4]. Moreover, in this particular case, an additional problem is the in vivo racemisation of (R)-thalidomide into the (S)-thalidomide. So, even if the drug had been commercialised as a single enantiomer, it would have been risky because of racemisation process. Thus, acceding enantiomerically pure compounds in the development of pharmaceuticals, agrochemicals and flavours is a very significant endeavour, which has been stressed by the US Food and Drug Administration [5].
Among the well-known methods for asymmetric synthesis, catalysis gained an ever-growing success since it presents many advantages with respect to the traditional wasteful stoichiometric methods (chiral auxiliaries, chiral-pool approaches): the catalyst is used at low or very low loading, the starting material does not need to come from the chiral pool, and the synthesis does not require extra steps for the installation and removal of a chiral auxiliary. For these reasons, the chiral catalyst approach has been extensively exploited in the last decades and many review papers have been written on this topic [6–9]. Furthermore, the chemistry community has strongly believed in the potential of metal-catalysed transformation, which therefore has been broadly explored through the years leading to remarkable results in the stereoselective synthesis [10, 11]. Nevertheless, the use of metal-containing catalysts in homogeneous phase has some drawbacks, such as the difficulties and the costs related to the recovering the catalyst. Such disadvantages are overcome heterogeneous catalysis, in which the metal site is supported on or inserted into a solid. The use of enzymes has many attractive features in the context of the Green Chemistry approach, such as limited stability of enzymes under certain organic reaction conditions and low efficiency when using wild-type strains [12]. Furthermore, the use of high-purity enzymes and, often, of costly co-factors, can be a major shortcoming, thus limiting its effective scale-up at commercial level. A more recently developed approach to asymmetric catalysis, based on organic molecules, overcomes some of the issues related to metal-based catalysts and some difficulties connected to the use of enzymes.
DEFINITION AND ORIGINS OF ORGANOCATALYSIS
The word Organocatalysis has been introduced in the scientific community in 2000 by MacMillan, one of the pioneers of the field [13, 14]. Organocatalysis refers to the use of small organic molecules to catalyse organic transformations. During the last decades, organocatalysis has been included among the most important and successful concepts in asymmetric catalysis and it has been used for the enantioselective construction of C-C, C-N, C-O, C-S, C-P and C-halide bonds [15–20]. Furthermore, this new branch experienced a boom in the first eight years from its advent. The publication number conserved the initial trend in the last years and is still growing, with more than 2000 manuscripts published in the field, in which more than 150 reaction types are reported (Figure 2) [21]. In one of his reference work, MacMillan proposed that this big power of attraction could be attributed to the name organocatalysis itself, which provided a strong identity and helped to unify the field [22].

The history of organocatalysis has the special characteristic of being started more than a hundred years ago and actually being ignored as such an important and independent research field until the year 2000. In fact, in 1912, Bredig and Fiske reported that cinchona alkaloid promoted the addition of hydrogen cyanide to benzaldehyde with low enantioselectivity [23] and in 1960, Pracejus found a cinchona alkaloid catalysed asymmetric addition of methanol to ketenes [24]. Later, in 1971, Hajos's and Wiechert's groups reported independently the (S)-proline-catalysed asymmetric aldol reaction, without a fully satisfying mechanistic study and without recognising the important novelty of this finding (Figure 3) [25]. It was only in 2000, that Benjamin List reported an asymmetric (S)-proline (5) catalysed aldol reaction and officially kicked-off to the “golden age” of Organocatalysis, which is the leading topic of this short review.

ORGANOCATALYSIS AND NON-COVALENT INTERACTIONS
Activation modes and some exemplar outcomes
Among the various organocatalytic methodologies, there are different types of activation and two main areas can be identified on the mechanistic basis: (i) covalent organocatalysis and (ii) non-covalent organocatalysis [26].
In the former case, within the catalytic cycle, the catalyst covalently binds the substrate, in the latter case only non-covalent interactions, such as hydrogen bonding (H-bonding) or the formation of ion pairs, activate the molecule towards the asymmetric transformation (Figure 4) [27].

With this brief review contribution, we would like to give a sketchy overview on both types of activation modes, subsequently focusing on some recent outcomes in the field of non-covalent organocatalysis.
Covalent organocatalysis
In Covalent organocatalysis, it has been ascertained that the catalyst covalently binds the substrate, as depicted in the examples (a) and (b) presented in Figure 4. In the enamine catalysis, a catalytically generated enamine intermediate is formed via deprotonation of an iminium ion and it reacts with various electrophiles or undergoes pericyclic reactions. Typical examples are proline-catalysed aldol reactions which proceed via enamine intermediates, such as the one reported by List in 2000. The hypothesised mechanism involves the formation of the enamine between (S)-proline and acetone; (S)-proline is in fact able to increase the HOMO energy, and thus the reactivity, of the nucleophile. Specifically, in the reaction intermediate, the catalyst interacts with the aldehyde through the protonation of the carbonyl, leading to a deep stereocontrol. This pioneering reaction allowed the attainment of highly enantioenriched aldols in very good yield (Figure 5) [28].

Another example of catalysis based on the enamine mechanism, is the asymmetric synthesis of a high added value, such as oseltamivir (commercially also known as Tamiflu), which is based on simple starting materials, such as alkoxyaldehyde 12, nitroalkene 14 and diethyl vinylphosphonate derivative 15 (Figure 6) [29]. The first reaction, catalysed by the trimethylsilyl-protected prolinol 11, is the organocatalytic enantioselective reaction between 12 and 14, giving a Michael adduct. This adduct then reacts in the next step with 15 to give, after a Wittig reaction and a thio-Michael reaction, the intermediate 16, from which enantiopure oseltamivir 17 is achieved in 57% overall yield. This reaction sequence proceeds by three distinguished one-pot operations and only one column chromatography purification and it is the most efficient synthesis of oseltamivir reported so far [30].

An additional noteworthy application of enamine catalysis is the first catalytic asymmetric α-alkylation of α-branched aldehydes developed by List et al. starting from simple racemic α-branched aldehydes and achieving the corresponding enantiomerically enriched products, in a dynamic kinetic asymmetric transformation [31]. In this reaction, racemic aldehydes are converted into corresponding enantioenriched alkylated products in high yields, up to 80 % (Figure 7a). Furthermore, the reaction features a new sterically demanding proline-derived catalyst (18), which the authors hypothesise to be exploited in other asymmetric transformations in the future.

In iminium catalysis, the active species is an iminium ion formed by the reversible reaction of an amine catalyst with a carbonyl substrate. The higher reactivity of the iminium ion compared to the carbonyl species is utilised to facilitate reactions such as Knoevenagel condensations, cyclo- and nucleophilic additions and cleavage of σ-bonds adjacent to the α-carbon.
In 2000, MacMillan presented a direct Diels–Alder reaction catalysed by a new chiral imidazolidinone catalyst (22). This catalyst is able to direct the addition of a diene to enals, by lowering the energy of the lowest unoccupied molecular orbital, LUMO, thus activating the dienophile towards the diene addition (Figure 7b) [14]. These organocatalytic reactions paved the way to many research teams that started to work in the same field, thus developing many different catalysts types and contributing to the growth of the application potential for organocatalysis.
For the enantioselective epoxidation of α and β-branched enals, Hayashi's and Jørgensen's groups independently disclosed the remarkable potential of α,α-diaryl prolinol silyl ethers (30 and 31, Figure 8a and 8b), obtaining the desired epoxide in high enantioselectivities (up to 98% ee) and excellent yields (up to 90%) [32, 33]. Furthermore, both groups investigated on the reaction mechanism, hypothesising an iminium ion covalent catalysis. In fact, within the catalytic cycle, the key intermediate is an iminium ion (I), which lowers the LUMO energy of the electrophilic double bond of substrate. The subsequent stereoselective addition of the hydroperoxide anion, followed by the cyclisation, leads to the enantioenriched product (Figure 8c) [34]. The understanding of the mechanisms for covalent organocatalysis is growing and an ever-increasing number of evidences highlight that the reactivity in organocatalytic reactions is often influenced and modulated by secondary non-covalent interactions [35].

Non-covalent organocatalysis
Non-covalent organocatalysis has recently been explored by many research groups and a vigorous development in this area has been achieved as well [36]. This kind of catalysis is commonly attributed to either hydrogen bond (H-bond) network or ion pairs' formation.
To be more precise, as recently elucidated by Jacobsen, many different interactions are often cooperating within the reaction mechanism in order to lead to high enantioselectivities [37]. The known non-covalent interactions scope includes charge–charge, charge–dipole, dipole–dipole, charge-induced dipole, dipole-induced dipole forces, steric repulsion and H-bond (Figure 9).

Indeed non-covalent interactions are weaker and less directional with respect to covalent bonding. Thus, in order to get the same performance in terms of stereoselectivity, more than one interaction is requested in order to have an effective spatial constraints. In fact, this kind of catalysts enclose more than one functionality, such as H-bond donor or acceptors or acid/basic sites, capable of generating multiple weak interactions, although operating in a synergistic way. For this reason, many of such catalysts are also called bifunctional. Noteworthy examples of bifunctional non-covalent organocatalysts are the Soós thiourea (32) and diphenyl prolinol (33), depicted in Figure 10 [38]. Both of these catalysts bear H-bond donor groups and a Brønsted base functionality [39]. The different functionalities are supposed to cooperate within the reaction mechanisms in many cases, activating both electrophile [through the Lewis acid (LA)] and nucleophile (through the Brønsted base) (Figure 10) [40–42].

Explicit H-bonding organocatalysis
One refers to explicit H-bonding organocatalysis when the activation mode clearly relies on H-bond interactions, even though other secondary interactions may usually cooperate with them. For instance, Jacobsen's and Schreiner's thioureas (34, 35, Figure 11) are paradigmatic cases for this class of organocatalysts [43, 44]. They clearly resemble the enzymatic pockets found in serine proteases for the activation of the amide carbonyl towards the serine nucleophilic attack. Within what is called the “oxyanion hole”, the amide carbonyl is coordinated and activated by two NH groups, hence favouring the serine nucleophilic attack with the formation of a tetrahedral intermediate stabilised by H-bonding (Figure 11) [45, 46]. A variety of electrophiles can be activated towards nucleophilic asymmetric addition with the same action mode and a series of reactions have been investigated so far [47, 48]. Moreover, H-bonding catalysts can promote enantioselective reactions by stabilising anionic fragments, as in Claisen rearrangement, by binding an anion, as in Pictet–Spengler cyclisation and hydrocyanation of imines (Figure 12) [49–51]. In all of these cases, very good enantioselectivities are reported (up to 97% ee for instance in the Pictet–Spengler), thus verifying the potentialities of H-bond.


Regarding the classification of H-bonding catalysts within the Lewis/Brønsted acid/base catalysis branches, Schreiner and co-workers performed some pioneering studies concerning thioureas as H-bonding additives, by means of NMR, IR and computational techniques. The reactivity of these systems, in a Diels–Alder reaction, was also compared to the one recorded over two widely used LA (aluminium trichloride, titanium tetrachloride). These studies proved that many similarities are present between LA and H-bonding catalysts. This is the reason why some H-bonding catalysts can be classified as LA [38].
A noteworthy example was reported in 2011 by Shao and co-workers who has developed an organocatalytic asymmetric double Michael addition of nitroalkenes 52 to 3-substituted oxindoles 51. This transformation, catalysed by a thiourea catalyst (54) under mild conditions, leads to spirocyclopentaneoxindoles (53) with quite good diastereoselectivity and enantioselectivity (up to >30:1 dr; up to >99% ee) (Figure 13) [52]. The great potential of chiral thioureas has been recently employed in other asymmetric transformations [53, 54].

Another significant organocatalyst is the S-2-diphenylhydroxymethylpyrrolidine 33, which has been thoroughly exploited in organocatalysis. The first report about enantioselective catalytic reactions promoted by this chiral proline-derived molecule dates back to 2000 and it is applied to the epoxidation of alkenes. In the patent, it has been verified that the epoxidation of unfunctionalised alkenes can proceed efficiently and enantioselectively using prolinol 33 or other simple amines as catalysts and without the need for metal complex catalysts, in the presence of an oxidising agent [55]. This patent was followed by several papers. Some examples are enantioselective reductions [56], asymmetric hetero-Diels–Alder of aldehydes with enones [57] and self-aldol reaction, all of them being enamine-mediated [58]. Remarkably, diarylprolinols have shown promising performances in non-covalently activated reactions as well, and recent advances have been recently reviewed [59]. For instance, diphenylprolinol 33 and diarylprolinols 55 and 56 have been employed the enantioselective conjugate addition of N-heterocycles to α,β-enones (Figure 14a), and in the epoxidation of electron deficient olefins, such as enones acrylonitriles (Figure 14b), and useful building blocks such as α-ylidenoxindoles (Figure 14c) [60–64]. For the first organocatalytic addition of benzotriazole (58) to the enone 57, the authors hypothesise an H-bond network acting in the transition state towards the induction of a moderate enantioselectivity in the subsequent formation of the product 59. The group of Lattanzi, in the last decade achieved very good results in the epoxidation of electron deficient alkenes, such as enones and acrylonitriles, in addition to some elucidations on the reaction mechanism, which confirm the hypothesis of an H-bonding catalysis [65]. Furthermore, some interesting results have been achieved in the epoxidation of α-ylidenoxindoles, which are electron-poor alkenes bearing an oxindole core. This moiety is quite often retrieved in biologically active molecules, such as some anticancer agents. The organocatalytic epoxidation of such alkenes, through a non-covalent activation mode, in the presence of diphenylprolinol 33, as catalyst, and tert-butylhydroperoxide, TBHP, as oxidising agent, leads to the formation of enantioenriched spiroepoxy oxindoles (ee up to 82%, for compound 63). The reaction mechanism involves a transition state in which catalyst, substrate and oxidant are inserted in a hydrogen network, thank to the two H-bonding donor groups positioned on the catalyst, that are the amine and the hydroxyl.

Brønsted acid catalysis
Chiral Brønsted acids (BA) have in the last decades proved to be excellent organocatalysts, since they can catalyse a number of chemical transformations, leading to highly enantioenriched products.64 The substrate activation by BA is related to, though different from, H-bonding catalysis, since normally the formation of a H-bond precedes the transfer (partial or complete) of a proton. Moreover, some recent studies about BA catalysed imine activation suggest that besides ion pairing, due to the protonation of the substrate, H-bonding exists. This is a proof of the tight connection between H-bonding and BA catalysis. Before this study, it was assumed that full protonation of the imine resulted in the formation of an ion pair [66]. It is well known that BA could operate by means of specific or general acid catalysis [67, 68]. In specific BA catalysis, the electrophilic substrate is reversibly protonated in a pre-equilibrium step, prior to the nucleophile attack (II, Figure 15a). Conversely, in general BA catalysis, the proton is (partially or fully) transferred in the transition state of the rate-determining step (I, Figure 15a). In the activation of a compound, e.g. carbonylic derivative, towards chemical transformations, BA differ in the catalytic centre from the well-known LA. Indeed, in BA the centre is a proton instead of a metal and this goes along with the Green Chemistry principles (Figure 15b) [69]. However, proton has only one valence orbital, limiting the possible interactions. As a consequence, the BA catalyst scaffold should be sterically hindered in order to induce stereocontrol [70].
Chiral BA catalysis has had a breakthrough in asymmetric organocatalysis in 2004, when Terada and Akiyama independently presented an enantioselective Mannich-type reaction catalysed by new metal-free phosphoric acid, BINOL-derived compounds, such as 58 (Figure 16a) [71]. These pioneering studies showed how powerful could be the use of BA catalysis for asymmetric synthesis. BINOL-derived catalysts are designed in order to get an environment both sterically demanding and moderately acid, in order to achieve good enantioselectivity and to protonate the imine nitrogen [72]. While the first studies do not elucidate the specific mechanistic pathway (even though they suggest the protonation of the imide by the phosphoric acidic site), later on, the BA catalytic activity came up. Phosphoric acids bear an acidic site (OH, Figure 15c) and a basic site (P=O, Figure 15c) on the same acidic species, allowing the simultaneous activation of a nucleophile and of an electrophile. In the wake of the first encouraging attainments, in the last decade many different types of chiral phosphoric acids have been synthesised and employed in a broad scope of enantioselective reactions. It is in this context, that List and co-workers paid particular attention onto chiral BA catalysts, developing a wide set of chiral phosphoric acids, whose utility is well recognised. The (S)-TRIP (59, Figure 15c), a binaphtyl phosphoric acid, is the most relevant example of this series since it has been employed for a broad range of reactions showing excellent results in terms of enantioselectivity. Noteworthy examples are Povarov reactions, O-alkylations, Pictet–Spengler reactions or benzidine rearrangement [73–76]. Chiral BA have shown particularly successful performances in the acetalisation reaction [77–79]. In 2005, Antilla et al. reported about the amidation of imines, which consists of a N,N-acetalisation. The reaction, catalysed by catalyst 62, under mild conditions, is very efficient and selective, although with a narrow substrate scope (Figure 16b) [80]. A recent application is in the intermolecular allylic amination of allylic alcohols catalysed by a phosphoramide, which leads to a variety of optically active allylic amines in good yields and ee values up to 94% [81]. The paper highlights that the amination site is mainly determined by electronic effects and the reaction is supposed to occur at the carbon adjacent to the electron-rich aryl group owing to a carbocation intermediate involved in the process.

A fascinating application of chiral BA catalysis is the synthesis of helicenes applying the Fischer indolisation (Figure 16c) [82]. The helical chirality strongly attracts attention in very diverse fields, such as material sciences, catalysis and biology. The authors could induce the stereochemistry of the helicene by means of a peculiar catalyst 63, derived from SPINOL ((S)-1,1′-spirobiindane-7,7′-diol) which bears extended π-substituents interacting with the substrate via π–π interactions [83, 84].

In 2012, List et al. developed a new concept: confined BA, extremely sterically demanding and bearing a chiral pocket that resemble the one found in enzymes [85]. These catalysts have been initially used for the asymmetric spiroacetalisation of sterically non-hindered compounds, in order to obtain biologically relevant spiroacetals, such as olean (major sex pheromone of the olive fruit fly; Figure 17), with very high enantiomeric excesses (96% ee). The new imidophosphoric acid still bears the double functionality, acid and base, on the two phosphates, allowing small and unhindered molecules to enter the active pocket and to be activated in such a peculiar environment towards the chemical transformation. This pioneering work paved the way to other enantioselective transformations, such as sulfoxidation and acetalisation, verifying further the potential of confined BA [86, 87].

HETEROGENISATION OF ORGANOCATALYSTS
One of the drawbacks of organocatalysis is that catalyst recovery and reuse is difficult. In the last years, envisaging some industrial uses for organocatalysts, a few attempts of anchoring them over solid supports have been made. In order to implement the concepts of Green Chemistry, the heterogeneous catalysts should be recyclable, selective, robust, cheap and more active than their homogeneous analogues [88]. However the anchoring of active homogeneous catalysts on solid supports may lead to less selective species. A fascinating example in this context is a recent invention by Lee et al., which makes use of nylon textiles as supports for organocatalysts [89, 90]. These new catalysts, OrganoTexCat (Figure 18), have been synthesised starting from the corresponding organocatalyst, exploiting the production of radicals on the nylon surface, induced by the irradiation under ultraviolet light. OrganoTexCat displays excellent stability, activity and recyclability and has been used for some organic transformations. For instance, the silylated glutamic anhydride (a valuable statin derivatives precursor) has been converted into compound 72, in a gram scale, in excellent yield and selectivity. The reaction has been performed by means of a continuous iterative reaction, which was performed in a continuous circle reactor, equipped with a column packed with the OrganoTexCat 70.

Another noteworthy example of supported organocatalyst is the one developed by Raja and co-workers [91]. They have illustrated the covalent heterogenisation of two homogeneous organocatalysts, cinchonine and 1,4-diazabicyclo[2.2.2]octane, onto the inner walls of mesoporous silica supports. This resulted in highly active, selective, recoverable solid catalysts, which were employed in the Michael addition reaction depicted in Figure 19. Some other successful examples of enantioselective transformations by immobilised organocatalysts are reported in the very recent literature [92–95].

CONCLUDING REMARKS
What lies ahead? From the number of publications, it is possible to affirm that organocatalysis is not experiencing a slowdown, presumably because many scientists have realised that its peculiarities and benefits can be exploited further and even enhanced. Many patents have already been disclosed on enantioselective organocatalytic transformations and this is a clear clue of the relevant potential of the technique [96–99]. Furthermore, a few organocatalytic procedures have recently been extended to industrial plants, even though the scale-up of a reaction is not straightforward [100]. For instance, the well-known L-proline-catalysed intramolecular aldol reaction (i.e., the Hajos–Parrish–Sauer–Wiechert process, see Figure 3, Chap. 1) has been implemented by two companies, Schering and Hoffman-La Roche for the asymmetric synthesis of steroids on multi-kilogram scale [101, 102]. We could cite many other examples of industrial applications of homogeneous organocatalysis, even though still some industrial manufacturers are reluctant in considering it as an alternative to classical methods [103].
However, it is envisaged that the immobilisation of a wide scope of organocatalysts will follow soon, with improvements in terms of recyclability of the catalyst as well as sustainability of the process and will help the fulfilment of a broader industrial exploitation. Indeed, thanks to a trans-disciplinary approach, merging the know-how in the field of catalysis, organic synthesis, materials science and macromolecular chemistry, it is possible to immobilise spatially confined highly selective organocatalysts, in order to obtain effective and stable artificial analogues of enzymes that are free from transition-metal active centres. This paves the way to enantioselective enzymomimetic catalysts with an enhanced robustness and with a better capability to withstand the severe reaction conditions of conventional industrial synthesis, with respect to their counterparts from biological origin.