- Record: found
- Abstract: found
- Article: found
Close Relative of Human Middle East Respiratory Syndrome Coronavirus in Bat, South Africa
letter
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
To the Editor: The severe acute respiratory syndrome (SARS) outbreak of 2002–03 and
the subsequent implication of bats as reservoir hosts of the causative agent, a coronavirus
(CoV), prompted numerous studies of bats and the viruses they harbor. A novel clade
2c betacoronavirus, termed Middle East respiratory syndrome (MERS)–CoV, was recently
identified as the causative agent of a severe respiratory disease that is mainly affecting
humans on the Arabian Peninsula (
1
). Extending on previous work (
2
), we described European Pipistrellus bat–derived CoVs that are closely related to
MERS-CoV (
3
). We now report the identification of a South Africa bat derived CoV that has an
even closer phylogenetic relationship with MERS-CoV.
During 2011–2012, fecal pellets were collected from 62 bats representing 13 different
species in the KwaZulu-Natal and Western Cape Provinces of South Africa and stored
in RNAlater solution (Life Technologies, Carlsbad, CA, USA). Details about the bat
sample are available in the Technical Appendix. RNA was extracted by using the QIAamp
Viral RNA Mini Kit (QIAGEN, Hilden, Germany). Screening for CoVs was done by nested
reverse transcription PCR using broadly reactive oligonucleotide primers targeting
a conserved region in the RNA-dependent RNA polymerase (RdRp) gene (online Technical
Appendix). PCR results were positive for 5 (8%) of the 62 specimens. PCR amplicons
for 4 positive specimens yielded alphacoronavirus sequences related to recently described
bat alphacoronaviruses from South Africa (
4
). The other positive specimen, termed PML/2011, was from an adult female Neoromicia
cf. zuluensis bat sampled in 2011; the specimen yielded a novel betacoronavirus (GenBank
accession no. KC869678). Technical Appendix Figure 1 shows the distribution of this
bat species.
To obtain better phylogenetic resolution, we extended the 398-nt RdRp fragment generated
by the screening PCR to 816 nt, as described (
5
). PML/2011 differed from MERS-CoV by only 1 aa exchange (0.3%) in the translated
816-nt RdRp gene fragment. Thus, PML/2011 was much more related to MERS-CoV than any
other known virus. The amino acid sequence of the next closest known relatives of
MERS-CoV, from European Pipistrellus bats (
3
), differed from MERS-CoV by 1.8%. The amino acid sequences of viruses from Nycteris
bats in Ghana (
3
) and the 2c prototype bat CoVs, HKU4 and HKU5, from China (
6
) differed by 5.5%–7.7% from MERS-CoV. The smaller 152- to 396-nt RdRp fragments of
2c bat CoVs from a Hypsugo savii bat in Spain (
7
), bat guano in Thailand (
8
), and a Nyctinomops bat in Mexico (
9
) showed no or only partial overlap with the 816-nt fragment generated in this study;
thus, a direct comparison could not be done. However, in their respective RdRp fragments,
these CoVs yielded amino acid sequence distances of 3.5%–8.0% and were thus probably
more distant from MERS-CoV than the virus described here.
A Bayesian phylogenetic analysis of the 816-nt RdRp sequence confirmed the close relationship
between PML/2011 and MERS-CoV (Figure). Their phylogenetic relatedness was as close
as that of SARS-CoV and the most closely related bat coronavirus known, Rs672 from
a Rhinolophus sinicus bat (Figure). Like PML/2011 and MERS-CoV, Rs672 and SARS-CoV
showed only 1 aa exchange in the translated 816-nt RdRp fragment. To confirm this
relatedness, we amplified and sequenced a short 269-nt sequence encompassing the 3′-terminus
of the spike gene for PML/2011 (oligonucleotide primers available upon request from
the authors). A partial spike gene–based phylogeny using this sequence yielded the
same topology as that using the partial RdRp sequence (Technical Appendix Figure 2).
Again, PML/2011 was most closely related to MERS-CoV, showing only a 10.9% aa sequence
distance in this gene, which encodes the glycoprotein responsible for CoV attachment
and cellular entry. This distance was less than the 13.3% aa sequence distance between
MERS-CoV and the European Pipistrellus CoVs (
3
) and less than the 20.5%–27.3% aa sequence distance between MERS-CoV and HKU5 and
between MERS-CoV and HKU4 (
6
) in the same sequence fragment.
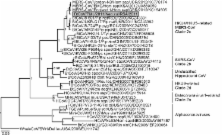
Figure
Partial RNA-dependent RNA polymerase (RdRp) gene phylogeny, including the novel betacoronavirus
from a Neoromicia zuluensis bat in South Africa (GenBank accession no. KC869678 for
both partial RdRp and spike gene sequences). The Bayesian phylogeny was done on a
translated 816-nt RdRp gene sequence fragment, as described (
5
). MrBayes V3.1 (http://mrbayes.sourceforge.net/) was used with a WAG substitution
model assumption over 2,000,000 generations sampled every 100 steps, resulting in
20,000 trees, of which 25% were discarded as burn-in. A whale gammacoronavirus was
used as an outgroup. The novel N. zuluensis bat virus is highlighted in gray. Values
at deep nodes represent statistical support from posterior probabilities. Only values
>0.9 are shown. Coronavirus clades are depicted to the right of taxa. Scale bar represents
genetic distance. MERS-CoV, Middle East respiratory syndrome coronavirus; SARS, severe
acute respiratory syndrome; Bt-CoV, bat coronavirus; HCoV, human coronavirus, MHV,
mouse hepatitis virus; FCoV, feline coronavirus; TGEV, transmissible gastroenteritis
coronavirus.
Our results further support the hypothesis that, like human CoV-229E and SARS-CoV,
ancestors of MERS-CoV might exist in Old World insectivorous bats belonging to the
family Vespertilionidae, to which the genera Neoromicia and Pipistrellus belong (
3
). Knowledge of the close relatedness of PML/2011 and MERS-CoV, which contrasts with
the more distant relatedness of CoVs in bats from the Americas and Asia, enables speculations
of an African origin for bat reservoir hosts of MERS-CoV ancestors. This hypothesis
is limited by a global sampling bias, the small sample size, and the single clade
2c betacoronavirus detection in this study. Still, a putative transfer of MERS-CoV
ancestors from Africa to the Arabian Peninsula would parallel the transfer of other
viruses (e.g., the exportation of Rift Valley fever virus from East Africa, which
led to a severe outbreak in Saudi Arabia in 2000) (
10
).
Studies of Vespertilionidae bats and potential intermediate hosts (e.g., carnivores
and ungulates, such as camels) are urgently needed to elucidate the emergence of MERS-CoV.
Such studies should focus on the Arabian Peninsula and Africa.
Technical Appendix
Description of bat sampling, screened bat species, distribution of Neoromicia zuluensis
bats, and spike gene phylogeny of the 2c betacoronavirus clade.
Related collections
Most cited references5
- Record: found
- Abstract: found
- Article: not found
Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia.
- Record: found
- Abstract: found
- Article: not found
Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features.
Patrick C. Y. Woo, Ming Wang, Susanna Lau … (2007)
- Record: found
- Abstract: found
- Article: found
Coronaviruses in bats from Mexico
S. Anthony, R Ojeda-Flores, O. Rico-Chávez … (2013)