- Record: found
- Abstract: found
- Article: found
Essential Roles of BCCIP in Mouse Embryonic Development and Structural Stability of Chromosomes
Read this article at
Abstract
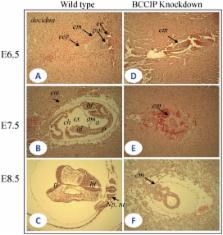
BCCIP is a BRCA2- and CDKN1A(p21)-interacting protein that has been implicated in the maintenance of genomic integrity. To understand the in vivo functions of BCCIP, we generated a conditional BCCIP knockdown transgenic mouse model using Cre-LoxP mediated RNA interference. The BCCIP knockdown embryos displayed impaired cellular proliferation and apoptosis at day E7.5. Consistent with these results, the in vitro proliferation of blastocysts and mouse embryonic fibroblasts (MEFs) of BCCIP knockdown mice were impaired considerably. The BCCIP deficient mouse embryos die before E11.5 day. Deletion of the p53 gene could not rescue the embryonic lethality due to BCCIP deficiency, but partially rescues the growth delay of mouse embryonic fibroblasts in vitro. To further understand the cause of development and proliferation defects in BCCIP-deficient mice, MEFs were subjected to chromosome stability analysis. The BCCIP-deficient MEFs displayed significant spontaneous chromosome structural alterations associated with replication stress, including a 3.5-fold induction of chromatid breaks. Remarkably, the BCCIP-deficient MEFs had a ∼20-fold increase in sister chromatid union (SCU), yet the induction of sister chromatid exchanges (SCE) was modestly at 1.5 fold. SCU is a unique type of chromatid aberration that may give rise to chromatin bridges between daughter nuclei in anaphase. In addition, the BCCIP-deficient MEFs have reduced repair of irradiation-induced DNA damage and reductions of Rad51 protein and nuclear foci. Our data suggest a unique function of BCCIP, not only in repair of DNA damage, but also in resolving stalled replication forks and prevention of replication stress. In addition, BCCIP deficiency causes excessive spontaneous chromatin bridges via the formation of SCU, which can subsequently impair chromosome segregations in mitosis and cell division.
Author Summary
BCCIP is a BRCA2- and p21-interacting protein. Studies with cell culture systems have suggested an essential role of BCCIP gene in homologous recombination and suppression of replication stress and have suggested that BCCIP defects causes mitotic errors. However, the in vivo function(s) of BCCIP and the mechanistic links between BCCIP's role in suppression of replication stress and mitotic errors are largely unknown. We generated transgenic mouse lines that conditionally express shRNA against the BCCIP, and we found an essential role of BCCIP in embryo development. We demonstrate that BCCIP deficiency causes the formation of a unique type of structural abnormality of chromosomes called sister chromatid union (SCU). It has been noted in the past that impaired homologous recombination and resolution of stalled replication forks can have detrimental consequences in mitosis. However, the physical evidence for this link has not been fully identified. SCU is the product of ligation between sister chromatids, likely formed as a result of unsuccessful attempt(s) to resolve stalled replication forks. Because the SCU will progress into chromatin bridges at anaphase, resulting in mitosis errors, it likely constitutes one of the physical links between S-phase replication stress and mitotic errors.
Related collections
Most cited references44
- Record: found
- Abstract: found
- Article: not found
Tumor spectrum analysis in p53-mutant mice.
- Record: found
- Abstract: found
- Article: not found
Efficient in vivo manipulation of mouse genomic sequences at the zygote stage.
- Record: found
- Abstract: found
- Article: not found