- Record: found
- Abstract: found
- Article: found
Revisiting post-infectious glomerulonephritis in the emerging era of C3 glomerulopathy

Read this article at
Abstract
Background
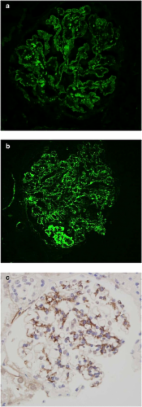
Post-infectious glomerulonephritis (PIGN) is an immune complex-mediated glomerular injury that typically resolves. Dominant C3 deposition is characteristic of PIGN, but with the emergence of C3 glomerulonephritis (C3GN) as a distinct entity, it is unclear how the pathologic similarities between PIGN and C3GN should be reconciled. Therefore, nephrologists and nephropathologists need additional guidance at the time of biopsy.
Methods
We studied 23 pediatric and young adult patients diagnosed with PIGN. Patients were divided into two groups, one with co-dominance between C3 and immunoglobulins and the other meeting proposed diagnostic criteria for C3GN. Clinical and pathological features were compared.
Results
No clinical and/or pathological features could distinguish between those with C3-co-dominant deposits and those with C3 dominance. Nearly all patients in both groups regained their baseline renal function without clinical intervention.
Conclusions
Although the identification of abnormalities of the alternative pathway of complement is characteristic of C3GN, testing is not widely available and the turnaround time often exceeds 1 month. Our study found that PIGN with either co-dominant or dominant C3 deposition in a cohort of young patients has excellent short-term outcomes. Close clinical observation for persistent abnormalities, such as hypocomplementemia, prolonged hematuria or proteinuria, is recommended to single out patients that may harbor intrinsic complement abnormalities.
Related collections
Most cited references16
- Record: found
- Abstract: found
- Article: not found
C3 Glomerulonephritis: Clinicopathologic findings, complement abnormalities, glomerular proteomic profile, treatment and follow-up
- Record: found
- Abstract: found
- Article: not found