- Record: found
- Abstract: found
- Article: found
KCa3.1 mediates dysfunction of tubular autophagy in diabetic kidneys via PI3k/Akt/mTOR signaling pathways
Read this article at
Abstract
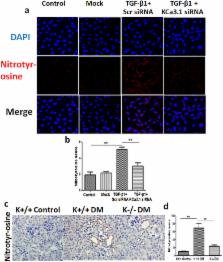
Autophagy is emerging as an important pathway in many diseases including diabetic nephropathy. It is acknowledged that oxidative stress plays a critical role in autophagy dysfunction and diabetic nephropathy, and KCa3.1 blockade ameliorates diabetic renal fibrosis through inhibiting TGF-β1 signaling pathway. To identify the role of KCa3.1 in dysfunctional tubular autophagy in diabetic nephropathy, human proximal tubular cells (HK2) transfected with scrambled or KCa3.1 siRNAs were exposed to TGF-β1 for 48 h, then autophagosome formation, the autophagy marker LC3, signaling molecules PI3K, Akt and mTOR, and oxidative stress marker nitrotyrosine were examined respectively. In vivo, LC3, nitrotyrosine and phosphorylated mTOR were examined in kidneys of diabetic KCa3.1+/+ and KCa3.1−/− mice. The results demonstrated that TGF-β1 increased the formation of autophagic vacuoles, LC3 expression, and phosphorylation of PI3K, Akt and mTOR in scrambled siRNA transfected HK2 cells compared to control cells, which was reversed in KCa3.1 siRNA transfected HK2 cells. In vivo, expression of LC3 and nitrotyrosine, and phosphorylation of mTOR were significantly increased in kidneys of diabetic KCa3.1+/+ mice compared to non-diabetic mice, which were attenuated in kidneys of diabetic KCa3.1−/− mice. These results suggest that KCa3.1 activation contributes to dysfunctional tubular autophagy in diabetic nephropathy through PI3K/Akt/mTOR signaling pathways.
Related collections
Most cited references30
- Record: found
- Abstract: found
- Article: not found
The thioredoxin system as a therapeutic target in human health and disease.
- Record: found
- Abstract: found
- Article: not found
Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences.

- Record: found
- Abstract: found
- Article: found