- Record: found
- Abstract: found
- Article: found
OL-EDA-ID Syndrome: a Novel Hypomorphic NEMO Mutation Associated with a Severe Clinical Presentation and Transient HLH
letter
Silvia Ricci
1
,
,
Francesca Romano
1 ,
Francesco Nieddu
1 ,
Capucine Picard
2
,
3
,
4
,
5 ,
Chiara Azzari
1
12 November 2016
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
To the Editor:
Background
NEMO deficiency syndrome is a rare and heterogeneous condition that presents in early
infancy. The phenotypic spectrum is broad ranging from hypogammaglobulinemia and mild
ectodermal signs to OL-EDA-ID syndrome, the most severe phenotype for IKBKG hypomorphic
mutations [1].
NF-κB is a transcriptional factor involved in many signaling pathways, and NEMO plays
a key role in its activation. Many human hypomorphic mutations in IKBKG have been
described involving IL-1 family protein receptors, TLR, VEGFR-3, RANK, the ectodysplasin-A
receptor, CD40, and TNF receptor signals. It is known that the regulation of genes
essential for cell adhesion, cell survival, immunoglobulin class switching, osteoclast
function, and T and B cell development can be impaired but the knowledge of mechanisms
that explain the relationship between genotype and phenotype is still incomplete.
Case Report
We examined a 3-month-old infant with persistent diarrhea and failure to thrive. The
male patient was born at term to healthy non-consanguineous Italian parents after
an uncomplicated pregnancy. His umbilical separation was delayed. At admission, he
had a temperature of 38 °C, CRP concentration of 41 mg/L, white blood cell count of
34.6 × 103/μl, polymorphonuclear cell count of 14.11 × 103/μl, lymphocytic cell count
of 15.43 × 103/μl, and hypereosinophilia (3.8 × 103/μl). Adenovirus was detected in
his stool sample. Physical examination revealed mild dysmorphic features with frontal
bossing, micrognathia, hypoplastic nasal wings, and flattened alveolar margins. He
presented signs of anhidrotic ectodermal dysplasia with fine and sparse hair, absent
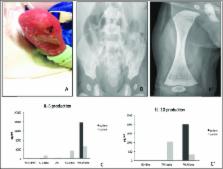
eyebrows, thin translucent skin with dry eczema, and hyperkeratosis (Fig. 1(A)). The
absence of sweat glands at skin biopsy confirmed this hypothesis. Radiologic evaluation
was performed and the images showed “bone-in-bone appearance” of the femoral, iliac,
and ischial bones bilaterally, consistent with a diagnosis of osteopetrosis (Fig. 1(B,
B′)). His general condition rapidly deteriorated with significant dyspnea, oliguria,
and lethargy. He presented acute seizures. A head CT scan was negative. MRI was not
performed. He was immediately admitted to the Pediatric Intensive Care Unit. On admission,
he had leukocytosis (52.24 × 103/μl) and the signs of secondary hemophagocytic lymphohistiocytosis
with prolonged fever, hepatosplenomegaly, anemia (Hb 6.9 g/dl), elevated ferritinemia
(37,300 ng/ml), LDH (2105 U/L), low fibrinogen (93 mg/dl), and hypertriglyceridemia
(700 mg/dl). Conventional respiratory and circulatory support with inotropes was necessary.
A thoracic scan image was concordant with interstitial pneumonitis. Pneumocystis jirovecii
and CMV DNA were identified in the bronchoalveolar lavage by PCR. The patient was
successfully treated with meropenem, trimethoprim/sulfamethoxazole, and ganciclovir.
The signs of concomitant macrophage activation syndrome were gradually normalized
under systemic corticosteroid therapy. During his prolonged hospitalization, the patient
also displayed a transient and inconstant lymphedema of the lower limbs. Immunological
assay was performed at 4 months of life. The patient presented severe hypogammaglobulinemia
with IgG (1.73 g/L with normal range 2.22–8.46 g/L), IgA (0.01 g/L with normal range
0.06–0.6 g/L), and IgM (0.04 g/L with normal range 0.28–0.39 g/L). TRECs and KRECs
were normal. Flow cytometry immunophenotyping revealed low NK and B memory cell counts
(Table 1). NK cell functional activity (CD107a expression) could not be determined
due to the low number of NK cells. Proliferative T cell responses to mitogenic stimuli
were normal (Table 1). We performed an analysis of the IKBKG sequence (NM_001099856.3)
from both genomic DNA and cDNA and we identified a novel missense mutation c.1238A>G
(p.H413R) within exon 10 on the zinc finger domain. The substituted histidine is highly
conserved among different species (Fig. 2). Using a bioinformatic system (Polyphen
and SIFT-Sort Intolerant From Tolerant), the new mutation is predicted to be damaging
with a score of 0.99 (sensitivity 0.09; specificity 0.99) and to affect protein function
with a score of 0.00 (intolerant), respectively. The CADD score was also elevated
(22.9). We performed a careful dermatological examination of the patient’s mother,
we did not observe any of the nail, hair, dental, or skin findings typical of incontinentia
pigmenti. Moreover, there was no history of dermatological problems in the family.
Maternal sequencing on genomic and cDNA revealed the WT IKBKG on both alleles. We
evaluated the patient’s blood cells after activation with TNF-α, IL-1β, and other
agonists of TLRs. The response of the patient’s blood cells to IL-1β, LPS (agonist
of TLR-4), and SAC was abnormal, in terms of IL-6 production (Fig. 1(C)). Moreover,
the response to TNF-α was impaired in terms of IL-10 production (Fig. 1(C′)). IL-6
and IL-10 production was measured by enzyme-linked immunosorbent assay (ELISA) after
48 h of activation. For persistent bloody diarrhea, hypoproteinemia and feeding intolerance
parenteral nutrition was necessary. Colon biopsy showed macroscopic signs of enterocolitis
with diffuse eosinophilic infiltrates in the lamina propria of the colon. Despite
IV antimicrobial prophylaxis and regular infusions of IV immunoglobulin, at 10 months
of age, the patient was readmitted presenting a new episode of catheter-associated
bacteremia from E. coli. At 13 months of age, the patient underwent a myeloablative
conditioning regimen consisting of thiotepa, treosulfan, and fludarabine followed
by haploidentical stem cell transplantation with TCR-alpha/beta and CD19 depletion.
He received anti-thymocyte globulin and mycophenolate mofetil with a complete antimicrobial
prophylaxis as a prevention of acute GVHD and infectious complications. He died unexpectedly
from acute respiratory distress and sepsis due to multiresistant Pseudomonas aeruginosa
6 days post HSCT.
Fig. 1
Right lower extremity with signs of ectodermal dysplasia (A). Radiological findings
of osteopetrosis with increased bone intensity, bone-within-bone appearance of the
iliac and ischial bones (B), and of femoral epiphyses (B′). Functional test evaluation
performed by ELISA: impaired IL-6 production in patient after 48 h of activation of
whole blood with IL-1β, LPS, and S aureus Cowan I (SAC) (C) and impaired IL-10 production
after 48 h of activation with TNF-α. Increased IL-6 and IL-10 production after PMA/Ionomycin
stimulation (C′). The results (C and C′) are representative of two tests
Table 1
Patient’s lymphocyte phenotyping and T-lymphocytes proliferation in response to mitogens,
performed by FACS at 5 months of life
Cell population
%
Cells/μl
Lymphocytes
7945
T-Lymphocytes
75
7282
CD3+CD4+
66
6369
CD3+CD8+
9
881
CD45+CD3+CD4+CD31+
24
1528
CD45+CD3+CD4+CD45RO+
54
3439
CD45+CD3+CD4+CD45RA+
46
2930
CD45+CD3+CD4+CD45RA+CD31+
39
2485
B-Lymphocytes
5
481
CD27+
2
10
CD27+IgM+IgD+
76
9
CD27+IgM−IgD−
24
2
CD3−CD16+CD56+
2
206
CD3−CD19+CD40+
99
1906
CD3+CD8−CD40L
97
6175
T-lymphocytes proliferation assay
CFSE+ w/o stimulus
98%
PHA (10 μM)
77%
Anti-CD3 antibody and IL-2 (30 U/ml)
85%
Fig. 2
The NEMO gene structure and its zinc finger (ZF) which extends from amino acid residues
397 to 419. The three cysteine residues and the single histidine (H413) residue that
coordinate a zinc ion are indicated. H413 is high conserved among different species
(modified by Shifera AS The zinc finger domain of IKKγ (NEMO) protein in health and
disease J. Cell. Mol. Med.)
Discussion and Conclusion
We report a case characterized by a severe clinical presentation and fatal outcome
associated with a genotype never previously described. The clinical features of the
case, in particular the EDA signs and the life-threatening infection, led us to consider
a diagnosis of a severe form of NEMO deficiency syndrome. Indeed, the association
of osteopetrosis/lymphedema and EDA-ID is indicative of the most severe phenotype.
The immunological assay was completely compatible with previously reported cases.
As previously described, the CD40-CD40L signaling on the dendritic cells and B cells
involves NEMO in NF-κB activation. Impaired CD40 signaling in pulmonary dendritic
cells may result in a major susceptibility to fungi infections similar to patients
with hyper IgM syndrome [2]. Moreover, the patient displayed a very low NK cell count
which can explain his increased susceptibility to the herpes virus group, including
CMV [3]. The use of HSCT in NEMO deficiency syndrome remains controversial and few
data are available concerning the transplantation of a patient with a novel NEMO mutation
[5]. Despite the known intrinsic difficulties with engraftment for these patients,
having evaluated the severity of his life-threatening infections and their sequelae,
we decided that HSCT was the only therapeutic option. A similar case of OL-EDA-ID
syndrome caused by the substitution of a stop codon with a tryptophan (X420W) on the
zinc finger region in the NEMO gene has been reported. This substitution changes the
length of the final protein, resulting 27 amino acids longer than the WT protein [2].
Here, we describe a completely different genotype, a novel missense mutation, which
substitutes histidine for arginine at amino acid 413 on the zinc finger domain, and
which resulted in an equally severe phenotype. In addition, this case is characterized
by the precocious and serious development of intestinal failure and hemophagocytic
lymphohistiocytosis. These clinical signs have previously been described but never
together in association with a severe OL-EDA-ID phenotype. Recently, a variation involving
the same amino acid—p.H413Y—causing IP syndrome in a female patient with a random
X-inactivation has been reported [4]. In 2008, Cordier et al. presented a complete
analysis of the structural and functional properties of the ZF motif of NEMO. The
integrity of the tetrahedral zinc coordination site formed by H413 and another three
cysteine residues determines the ββα scaffold of NEMO ZF (Fig. 2). In our case, the
highly conserved H413 is substituted by an arginine; this implies an impaired stability
of the ZF fold which may alter its protein recognition abilities. It appears that
the disruption of C-terminus of NEMO gene, caused by nonsense mutation or by a missense
mutation of a key conjugating residue, lead to similar severe phenotype. Moreover,
functional complementation assays using the patient’s mononuclear cells showed that
the H413R NEMO mutation leads to a strong defect of LPS, IL-1β, and TNF-α-induced
NF-κB activation, as compared to WT NEMO (Fig. 1(C, C′)). Interestingly, cytokine
production is higher in patient than in control after PMA/ionomycin stimulation. Zilberman-Rudenko
et al. have recently described a distinct group of patient with inflammatory symptoms
caused by gain-of-function C-terminus deletion which confers increased responsiveness
to innate immune stimuli. Regulated activation of NF-kB transcription factors family
is important in immune cell function and inflammatory responses. Our case represents
an example of immunodysregulation characterized for clinical and molecular features
of immunodeficiency and auto-inflammation. We are aware that more studies are necessary
to establish all pathogenetic mechanisms of this point mutation; unfortunately, we
did not obtain the family’s consent for a second skin biopsy or for other investigations.
In conclusion, we report a novel missense mutation responsible for one of the most
complex and severe clinical presentations of reported NEMO deficiency cases. It appears
that the disruption of C-terminus of NEMO gene, caused by nonsense mutation or by
a missense mutation of a key conjugating residue, lead to similar severe phenotype.
We recommend that each novel variation is described and submitted to public databases
because any additional data will provide insight into genotype-phenotype correlation
and will improve patient care for infants with NEMO deficiency syndrome.
Related collections
Most cited references2
- Record: found
- Abstract: found
- Article: not found
Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity.
Eric Hanson, Linda Monaco-Shawver, Laura Solt … (2008)
- Record: found
- Abstract: found
- Article: not found