- Record: found
- Abstract: found
- Article: not found
Insertion Sequence–Driven Diversification Creates a Globally Dispersed Emerging Multiresistant Subspecies of E. faecium
Read this article at

Abstract
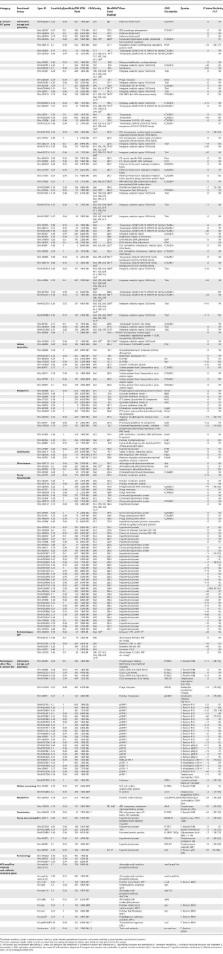
Enterococcus faecium, an ubiquous colonizer of humans and animals, has evolved in the last 15 years from an avirulent commensal to the third most frequently isolated nosocomial pathogen among intensive care unit patients in the United States. E. faecium combines multidrug resistance with the potential of horizontal resistance gene transfer to even more pathogenic bacteria. Little is known about the evolution and virulence of E. faecium, and genomic studies are hampered by the absence of a completely annotated genome sequence. To further unravel its evolution, we used a mixed whole-genome microarray and hybridized 97 E. faecium isolates from different backgrounds (hospital outbreaks ( n = 18), documented infections ( n = 34) and asymptomatic carriage of hospitalized patients ( n = 15), and healthy persons ( n = 15) and animals ( n = 21)). Supported by Bayesian posterior probabilities (PP = 1.0), a specific clade containing all outbreak-associated strains and 63% of clinical isolates was identified. Sequencing of 146 of 437 clade-specific inserts revealed mobile elements ( n = 74), including insertion sequence (IS) elements ( n = 42), phage genes ( n = 6) and plasmid sequences ( n = 26), hypothetical ( n = 58) and membrane proteins ( n = 10), and antibiotic resistance ( n = 9) and regulatory genes ( n = 11), mainly located on two contigs of the unfinished E. faecium DO genome. Split decomposition analysis, varying guanine cytosine content, and aberrant codon adaptation indices all supported acquisition of these genes through horizontal gene transfer with IS 16 as the predicted most prominent insert (98% sensitive, 100% specific). These findings suggest that acquisition of IS elements has facilitated niche adaptation of a distinct E. faecium subpopulation by increasing its genome plasticity. Increased genome plasticity was supported by higher diversity indices (ratio of average genetic similarities of pulsed-field gel electrophoresis and multi locus sequence typing) for clade-specific isolates. Interestingly, the previously described multi locus sequence typing–based clonal complex 17 largely overlapped with this clade. The present data imply that the global emergence of E. faecium, as observed since 1990, represents the evolution of a subspecies with a presumably better adaptation than other E. faecium isolates to the constraints of a hospital environment.
Author Summary
Whole-genome sequencing has become instrumental in investigating the genome contents of bacteria. However, there is enormous diversity within bacterial populations, and annotation of multiple genomes is costly and elaborate. For investigating diversity and phylogeny within bacterial species, comparative genomic hybridization is an attractive alternative that may provide fundamental insights into the factors (genes) distinguishing bacterial subpopulations. Enterococcus faecium, a worldwide emerging nosocomial pathogen usually resistant to multiple antibiotics, causes infections in immunocompromised patients. Using comparative genomic hybridization of 97 E. faecium strains isolated from different epidemiological niches worldwide, a subpopulation of E. faecium strains was identified that was associated with invasive infections and hospital outbreaks. Approximately 13% of the E. faecium pangenome was highly specific for this subpopulation, and, based on phylogenetic clustering, it should be considered a subspecies. We hypothesize that extensive variation within specific functional genes and high prevalence of mobile elements, mostly insertion sequence elements, contributed to the success of this genetic subset in its competition with other enterococci in hospital settings, creating a novel globally dispersed nosocomial subspecies. These findings fully confirmed previous phylogenetic studies based on multi locus sequence typing that had also revealed a genetic subset of E. faecium, clonal complex 17. Identification of genes specific for clonal complex 17 is a first step in elucidating how global spread and adaptation to the hospital environment of this emerging nosocomial pathogen has occurred.
Related collections
Most cited references83
- Record: found
- Abstract: not found
- Article: not found
Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing
- Record: found
- Abstract: found
- Article: not found
The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications.
- Record: found
- Abstract: found
- Article: not found