- Record: found
- Abstract: found
- Article: found
Activating transcription factor 3 modulates protein kinase C epsilon activation in diabetic peripheral neuropathy
Abstract
Background
Skin denervation that develops in patients with diabetes mellitus as a neuropathic manifestation is known as diabetic peripheral neuropathy (DPN). Skin denervation is parallel to neuronal injuries that alter intracellular signaling. To date, the correlation between nerve injury and the activation of intracellular responses to neuropathic manifestations has not been elucidated; specifically, whether activating transcription factor 3 (ATF3) is responsible for neuronal injury and a critical molecule that modulates the activation of intracellular protein kinase C epsilon (p-PKCε) and pain development in DPN is a crucial question.
Methods
To address, ATF3 knockout ( atf3 −/− group, C57/B6 genetic background) and wild-type mice ( atf3 +/+ group) received a single dose of streptozotocin (200 mg/kg) to generate a mouse model of DPN.
Results
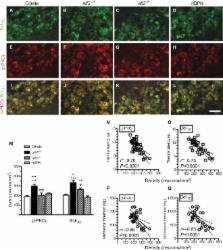
Both atf3 +/+ and atf3 −/− mice exhibited hyperglycemia and the same pathology of skin denervation at posttreatment month 2, but only atf3 +/+ mice developed thermal hyperalgesia ( P<0.001) and mechanical allodynia ( P=0.002). The atf3 +/+ group, but not the atf3 −/− group, had preferential ATF3 upregulation on p-PKCε(+) neurons with a ratio of 37.7%±6.1% in p-PKCε(+):ATF3(+) neurons ( P<0.001). In addition, B-cell lymphoma-extra large (Bcl- XL), an antiapoptotic Bcl2 family protein, exhibited parallel patterns to p-PKCε (ie, Bcl- XL upregulation was reversed in atf3 −/− mice). These two molecules were colocalized and increased by approximately two-fold in the atf3 +/+ group compared with the atf3 −/− group (30.0%±3.4% vs 13.7% ± 6.2%, P=0.003). Furthermore, linear analysis results showed that the densities of p-PKCε and Bcl- XL had a reverse linear relationship with the degrees of thermal hyperalgesia and mechanical allodynia.
Most cited references36
- Record: found
- Abstract: not found
- Article: not found
Ethical guidelines for investigations of experimental pain in conscious animals.
- Record: found
- Abstract: found
- Article: not found
Diabetic neuropathy: cellular mechanisms as therapeutic targets.
- Record: found
- Abstract: found
- Article: not found