- Record: found
- Abstract: found
- Article: found
MetaSort untangles metagenome assembly by reducing microbial community complexity
Read this article at
Abstract
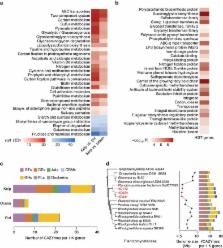
Most current approaches to analyse metagenomic data rely on reference genomes. Novel microbial communities extend far beyond the coverage of reference databases and de novo metagenome assembly from complex microbial communities remains a great challenge. Here we present a novel experimental and bioinformatic framework, metaSort, for effective construction of bacterial genomes from metagenomic samples. MetaSort provides a sorted mini-metagenome approach based on flow cytometry and single-cell sequencing methodologies, and employs new computational algorithms to efficiently recover high-quality genomes from the sorted mini-metagenome by the complementary of the original metagenome. Through extensive evaluations, we demonstrated that metaSort has an excellent and unbiased performance on genome recovery and assembly. Furthermore, we applied metaSort to an unexplored microflora colonized on the surface of marine kelp and successfully recovered 75 high-quality genomes at one time. This approach will greatly improve access to microbial genomes from complex or novel communities.
Abstract
Currently available metagenomic data analysis relies on reference genomes. Here, the authors describe a new de novo metagenomic assembly method, metaSort, that constructs bacterial genomes from metagenomic samples to reduce microbial community complexity while increasing genome recovery and assembly.
Related collections
Most cited references36
- Record: found
- Abstract: found
- Article: found
nhmmer: DNA homology search with profile HMMs
- Record: found
- Abstract: found
- Article: not found
Horizontal gene transfer, genome innovation and evolution.

- Record: found
- Abstract: found
- Article: found