- Record: found
- Abstract: found
- Article: found
The p53-p21-DREAM-CDE/CHR pathway regulates G 2/M cell cycle genes
Read this article at
Abstract
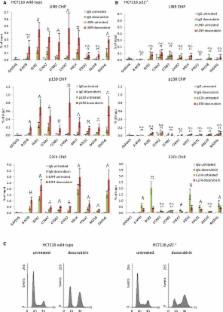
The tumor suppressor p53 functions predominantly as a transcription factor by activating and downregulating gene expression, leading to cell cycle arrest or apoptosis. p53 was shown to indirectly repress transcription of the CCNB2, KIF23 and PLK4 cell cycle genes through the recently discovered p53-p21-DREAM-CDE/CHR pathway. However, it remained unclear whether this pathway is commonly used. Here, we identify genes regulated by p53 through this pathway in a genome-wide computational approach. The bioinformatic analysis is based on genome-wide DREAM complex binding data, p53-depedent mRNA expression data and a genome-wide definition of phylogenetically conserved CHR promoter elements. We find 210 target genes that are expected to be regulated by the p53-p21-DREAM-CDE/CHR pathway. The target gene list was verified by detailed analysis of p53-dependent repression of the cell cycle genes B-MYB ( MYBL2), BUB1, CCNA2, CCNB1, CHEK2, MELK, POLD1, RAD18 and RAD54L. Most of the 210 target genes are essential regulators of G 2 phase and mitosis. Thus, downregulation of these genes through the p53-p21-DREAM-CDE/CHR pathway appears to be a principal mechanism for G 2/M cell cycle arrest by p53.
Related collections
Most cited references53
- Record: found
- Abstract: found
- Article: not found
The first 30 years of p53: growing ever more complex.
- Record: found
- Abstract: found
- Article: not found
p21 is necessary for the p53-mediated G1 arrest in human cancer cells.
- Record: found
- Abstract: found
- Article: not found