- Record: found
- Abstract: found
- Article: not found
Mycobacteria Attenuate Nociceptive Responses by Formyl Peptide Receptor Triggered Opioid Peptide Release from Neutrophils
Read this article at

Abstract
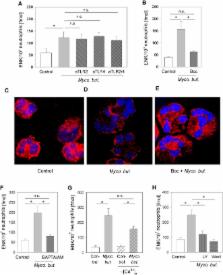
In inflammation, pain is regulated by a balance of pro- and analgesic mediators. Analgesic mediators include opioid peptides which are secreted by neutrophils at the site of inflammation, leading to activation of opioid receptors on peripheral sensory neurons. In humans, local opioids and opioid peptides significantly downregulate postoperative as well as arthritic pain. In rats, inflammatory pain is induced by intraplantar injection of heat inactivated Mycobacterium butyricum, a component of complete Freund's adjuvant. We hypothesized that mycobacterially derived formyl peptide receptor (FPR) and/or toll like receptor (TLR) agonists could activate neutrophils, leading to opioid peptide release and inhibition of inflammatory pain. In complete Freund's adjuvant-induced inflammation, thermal and mechanical nociceptive thresholds of the paw were quantified (Hargreaves and Randall-Selitto methods, respectively). Withdrawal time to heat was decreased following systemic neutrophil depletion as well as local injection of opioid receptor antagonists or anti-opioid peptide (i.e. Met-enkephalin, β-endorphin) antibodies indicating an increase in pain. In vitro, opioid peptide release from human and rat neutrophils was measured by radioimmunoassay. Met-enkephalin release was triggered by Mycobacterium butyricum and formyl peptides but not by TLR-2 or TLR-4 agonists. Mycobacterium butyricum induced a rise in intracellular calcium as determined by FURA loading and calcium imaging. Opioid peptide release was blocked by intracellular calcium chelation as well as phosphoinositol-3-kinase inhibition. The FPR antagonists Boc-FLFLF and cyclosporine H reduced opioid peptide release in vitro and increased inflammatory pain in vivo while TLR 2/4 did not appear to be involved. In summary, mycobacteria activate FPR on neutrophils, resulting in tonic secretion of opioid peptides from neutrophils and in a decrease in inflammatory pain. Future therapeutic strategies may aim at selective FPR agonists to boost endogenous analgesia.
Author Summary
Inflammation of peripheral tissue can be caused by bacteria and is frequently accompanied by pain. Pain severity depends on the balance of enhancing (proalgesic) and decreasing (analgesic) mediators. Local endogenous pain control involves the release of opioid peptides from immune cells at the site of inflammation. These opioid peptides bind to opioid receptors on peripheral nerves and inhibit transmission of nociceptive impulses. We hypothesized that bacteria can directly stimulate immune cells to release opioid peptides and thereby decrease pain. In a rat model, inoculation of the paw with heat-inactivated Mycobacterium butyricum led to local inflammation and pain responses. Nociceptive thresholds were further decreased (i.e. pain was enhanced) following immune cell (i.e. neutrophil) depletion, local injection of anti-opioid peptide antibodies or opioid receptor antagonists. Immune cells recognize bacteria by toll-like and/or formyl peptide receptors. Previous research indicated that mycobacteria enhance nociceptive responses via toll like receptors-2 and -4. We now demonstrate that mycobacteria also activate formyl peptide receptors on neutrophils leading to opioid peptide release and the inhibition of such responses. Since bacteria can simultaneously induce the generation of pro- and analgesic mediators, our results might be a further explanation for differences in pain between individual patients following bacterial infections.
Related collections
Most cited references75
- Record: found
- Abstract: not found
- Article: not found
Ethical guidelines for investigations of experimental pain in conscious animals.
- Record: found
- Abstract: found
- Article: not found