- Record: found
- Abstract: found
- Article: found
The Perchlorate Reduction Genomic Island: Mechanisms and Pathways of Evolution by Horizontal Gene Transfer
Read this article at
Abstract
Background
Perchlorate is a widely distributed anion that is toxic to humans, but serves as a valuable electron acceptor for several lineages of bacteria. The ability to utilize perchlorate is conferred by a horizontally transferred piece of DNA called the perchlorate reduction genomic island (PRI).
Methods
We compared genomes of perchlorate reducers using phylogenomics, SNP mapping, and differences in genomic architecture to interrogate the evolutionary history of perchlorate respiration.
Results
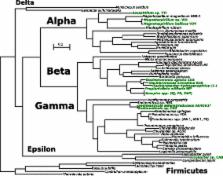
Here we report on the PRI of 13 genomes of perchlorate-reducing bacteria from four different classes of Phylum Proteobacteria (the Alpha-, Beta-, Gamma- and Epsilonproteobacteria). Among the different phylogenetic classes, the island varies considerably in genetic content as well as in its putative mechanism and location of integration. However, the islands of the densely sampled genera Azospira and Magnetospirillum have striking nucleotide identity despite divergent genomes, implying horizontal transfer and positive selection within narrow phylogenetic taxa. We also assess the phylogenetic origin of accessory genes in the various incarnations of the island, which can be traced to chromosomal paralogs from phylogenetically similar organisms.
Conclusion
These observations suggest a complex phylogenetic history where the island is rarely transferred at the class level but undergoes frequent and continuous transfer within narrow phylogenetic groups. This restricted transfer is seen directly by the independent integration of near-identical islands within a genus and indirectly due to the acquisition of lineage-specific accessory genes. The genomic reversibility of perchlorate reduction may present a unique equilibrium for a metabolism that confers a competitive advantage only in the presence of an electron acceptor, which although widely distributed, is generally present at low concentrations in nature.
Related collections
Most cited references40
- Record: found
- Abstract: found
- Article: not found
ProtTest: selection of best-fit models of protein evolution.
- Record: found
- Abstract: found
- Article: not found