- Record: found
- Abstract: found
- Article: found
Clinical and Genetic Characteristics of Chinese Patients with Occult Macular Dystrophy
Read this article at
Abstract
Purpose
To investigate the clinical and genetic characteristics of occult macular dystrophy (OMD) based on a Chinese patient cohort.
Methods
Fifteen Chinese OMD patients from nine unrelated families underwent genetic testing, and all of them harbored a pathogenic RP1L1 variant. Comprehensive ophthalmic examinations were performed in nine probands, including spectral-domain optical coherence tomography (SD-OCT), near-infrared reflectance (NIR), fundus autofluorescence (AF), and multifocal electroretinography.
Results
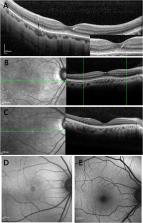
The RP1L1 variants p.R45W and p.S1199C were identified in 13 patients and two patients, respectively, and one was a de novo mutation. Among the nine probands, the median ages at onset and examination were 25.0 years (range, 6–51 years) and 27.0 years (range, 14–55 years), respectively. The median decimal visual acuity was 0.20 (range, 0.04–0.5). Foveal photoreceptor thickness and visual acuity showed a significant correlation ( r = 0.591; P = 0.01). All eyes presented with an absent interdigitation zone and blurred ellipsoid zone of photoreceptors when examined by SD-OCT. In addition, central round lesions with low NIR reflectance were observed in 66.7% (12/18) of eyes by NIR reflectance imaging, corresponding to the regions with abnormal photoreceptor microstructures observed by SD-OCT. Of the 18 eyes, only four eyes showed ring-like faint hyperfluorescence around the macula by AF.
Conclusions
To the best of our knowledge, this is the largest study in a cohort of Chinese OMD patients with RP1L1 mutations. Our findings revealed that the two recurrent RP1L1 variants are related to OMD in the Chinese population. Furthermore, multimodal imaging combined with genetic testing is valuable for diagnosing and monitoring OMD progression.
Related collections
Most cited references20
- Record: found
- Abstract: found
- Article: found
RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy.
- Record: found
- Abstract: found
- Article: not found
Dominant mutations in RP1L1 are responsible for occult macular dystrophy.
- Record: found
- Abstract: found
- Article: not found