- Record: found
- Abstract: found
- Article: found
PDL1 Regulation by p53 via miR-34
Read this article at
Abstract
Background:
Although clinical studies have shown promise for targeting PD1/PDL1 signaling in non–small cell lung cancer (NSCLC), the regulation of PDL1 expression is poorly understood. Here, we show that PDL1 is regulated by p53 via miR-34.
Methods:
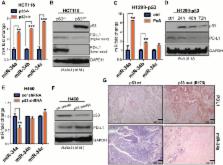
p53 wild-type and p53-deficient cell lines (p53 –/– and p53 +/+ HCT116, p53-inducible H1299, and p53-knockdown H460) were used to determine if p53 regulates PDL1 via miR-34. PDL1 and miR-34a expression were analyzed in samples from patients with NSCLC and mutated p53 vs wild-type p53 tumors from The Cancer Genome Atlas for Lung Adenocarcinoma (TCGA LUAD). We confirmed that PDL1 is a direct target of miR-34 with western blotting and luciferase assays and used a p53 R172HΔg/+K-ras LA1/+ syngeneic mouse model (n = 12) to deliver miR-34a–loaded liposomes (MRX34) plus radiotherapy (XRT) and assessed PDL1 expression and tumor-infiltrating lymphocytes (TILs). A two-sided t test was applied to compare the mean between different treatments.
Results:
We found that p53 regulates PDL1 via miR-34, which directly binds to the PDL1 3’ untranslated region in models of NSCLC (fold-change luciferase activity to control group, mean for miR-34a = 0.50, SD = 0.2, P < .001; mean for miR-34b = 0.52, SD = 0.2, P = .006; and mean for miR-34c = 0.59, SD = 0.14, and P = .006). Therapeutic delivery of MRX34, currently the subject of a phase I clinical trial, promoted TILs (mean of CD8 expression percentage of control group = 22.5%, SD = 1.9%; mean of CD8 expression percentage of MRX34 = 30.1%, SD = 3.7%, P = .016, n = 4) and reduced CD8 +PD1 + cells in vivo (mean of CD8/PD1 expression percentage of control group = 40.2%, SD = 6.2%; mean of CD8/PD1 expression percentage of MRX34 = 20.3%, SD = 5.1%, P = .001, n = 4). Further, MRX34 plus XRT increased CD8 + cell numbers more than either therapy alone (mean of CD8 expression percentage of MRX34 plus XRT to control group = 44.2%, SD = 8.7%, P = .004, n = 4). Finally, miR-34a delivery reduced the numbers of radiation-induced macrophages (mean of F4-80 expression percentage of control group = 52.4%, SD = 1.7%; mean of F4-80 expression percentage of MRX34 = 40.1%, SD = 3.5%, P = .008, n = 4) and T-regulatory cells.
Related collections
Most cited references35
- Record: found
- Abstract: found
- Article: not found
The blockade of immune checkpoints in cancer immunotherapy.
- Record: found
- Abstract: found
- Article: not found
Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade.
- Record: found
- Abstract: found
- Article: not found