- Record: found
- Abstract: found
- Article: found
Ultra wide field imaging of coats like response in Leber’s congenital amaurosis
brief-report

06 March 2017
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
A 12-year-old boy presented with poor vision and abnormal eye movements in his both
eyes since birth. There were no systemic complaints. The visual acuity was light perception
only in both eyes. The cycloplegic refraction revealed a hypermetropia of 6 and 7
diopters in right and left eye respectively, but the correction did not improve his
visual acuity. The child had pendular nystagmus; enophthalmos and oculo-digital sign
were present. The pupil reacted sluggishly to light in both the eyes. A dilated fundus
examination revealed bilateral optic disc pallor, pigment spicules all over the fundus
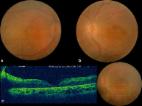
and vascular attenuation. In addition the right eye (Fig. 1) showed well-defined excavated
area of chorio-retinal atrophy in macula and sclerosed vessels with scattered retinal
hemorrhages in the temporal fundus. The left eye showed thick glial tissue with exudation
in macula (Fig. 2). An area of retinal vascular telangiectasias and subretinal exudation
was noted inferiorly and temporally in left fundus. Electroretinography (ERG) showed
extinguished scotopic and photopic responses. A diagnosis of Leber’s Congenital Amaurosis
(LCA) with “Coats like” response was made. Patient was advised for regular follow-up.
Figure 1
Ultra wide field pseudo colour image of the right eye showing disc pallor, wide spread
pigmentary changes, macular excavation and sclerosed vessels in temporal periphery.
Figure 2
Pseudo colour image of left eye showing disc pallor, pigmentary changes and fibro-glial
nodule at macula. In addition subretinal and retinal exudations along with telangiectasia
are seen in temporal and infero-temporal area.
Discussion
Leber’s congenital amaurosis is a severe hereditary retinal dystrophy that leads to
blindness manifesting in infancy. It is believed to be a cause of childhood blindness
in as many as 20% cases in blind institutes and accounts for 5% of all retinal dystrophies.
1
It is mostly inherited as an autosomal recessive trait and to date around 19 genes
have been implicated in the causation. Still, only around 70% or fewer cases are documented
to have the detailed genetic defects.
2
The fundus appearance in LCA can be extremely variable. Most cases initially present
with a normal fundus appearance or with mild granularity and vascular attenuation,
though in later life a number of changes have been described. Typical fundal changes
include macular coloboma, typical retinitis pigmentosa, retinitis punctata albescens,
optic atrophy, salt and pepper retinopathy, peripheral nummular pigmentation and vascular
complications, such as optic disc oedema, retinal vasculitis, secondary angiomatosis
and astrocytoma.
“Coats’ like response” is a clinical entity different from typical Coats disease and
can be seen in a variety of clinical scenarios. It is typically described as a vascular
alterations (telangiectasia and aneurysmal dilatation) with lipid exudation and has
been documented with conditions such pars planitis,
3
senile retinoschisis,
4
pigmented paravenous retinochoroidal atrophy,
5
linear en coup de sabre scleroderma
6
and retinitis pigmentosa (RP).7, 8, 9 Coats’ like response is relatively common in
RP associated with CRB1 mutation. Recently Hasan et al. described a case of LCA associated
with CRBI gene mutation, which had coats’ like response. Their patient had lesser
pigmentary changes, no macular coloboma and better visual acuity.
10
A definitive common pathogenic mechanism has not been described for such a response.
The clinical significance of such an observation arises from the fact that an exudative
response like this may add to the visual disability of the pre-existing illness by
extension of exudation to the posterior pole with ensuing macular oedema. In such
a case, treating the Coats’ like response may help mitigate some visual disability.
In our case we found LCA to be associated with macular exudation and mid-peripheral
telangiectasia associated with localized exudation. No intervention was performed
and the patient was advised for regular follow-up as there were no recent complaints
of decreased vision.
To our knowledge a “Coats’ like response” with LCA has never been documented before
with the help of ultra-wide field (UWF) imaging and may well be another clinical feature
in the already varied spectrum of fundus lesions associated LCA. UWF imaging is an
useful tool as it provides panoramic images of retina in a single click despite poor
fixation and cooperation from such patients.
Conflict of interest
The authors declared that there is no conflict of interest.
Related collections
Most cited references9
- Record: found
- Abstract: found
- Article: not found
Genetic testing for retinal dystrophies and dysfunctions: benefits, dilemmas and solutions.
Robert K Koenekoop, Rando Allikmets, Frans Cremers … (2007)

- Record: found
- Abstract: found
- Article: found
Coats-like retinitis pigmentosa: Reports of three cases
Emrah Kan, Turgut Yılmaz, Orhan Aydemir … (2007)
- Record: found
- Abstract: found
- Article: found
Coat’s like vasculopathy in leber congenital amaurosis secondary to homozygous mutations in CRB1: a case report and discussion of the management options
Somar Hasan, Arwa Azmeh, Osama Mostafa … (2016)