- Record: found
- Abstract: found
- Article: found
Performance of HADDOCK and a simple contact-based protein–ligand binding affinity predictor in the D3R Grand Challenge 2
Read this article at
Abstract
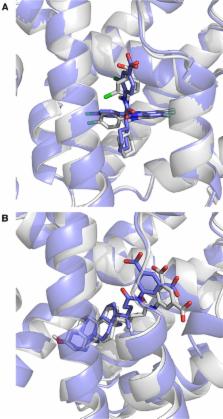
We present the performance of HADDOCK, our information-driven docking software, in the second edition of the D3R Grand Challenge. In this blind experiment, participants were requested to predict the structures and binding affinities of complexes between the Farnesoid X nuclear receptor and 102 different ligands. The models obtained in Stage1 with HADDOCK and ligand-specific protocol show an average ligand RMSD of 5.1 Å from the crystal structure. Only 6/35 targets were within 2.5 Å RMSD from the reference, which prompted us to investigate the limiting factors and revise our protocol for Stage2. The choice of the receptor conformation appeared to have the strongest influence on the results. Our Stage2 models were of higher quality (13 out of 35 were within 2.5 Å), with an average RMSD of 4.1 Å. The docking protocol was applied to all 102 ligands to generate poses for binding affinity prediction. We developed a modified version of our contact-based binding affinity predictor PRODIGY, using the number of interatomic contacts classified by their type and the intermolecular electrostatic energy. This simple structure-based binding affinity predictor shows a Kendall’s Tau correlation of 0.37 in ranking the ligands (7th best out of 77 methods, 5th/25 groups). Those results were obtained from the average prediction over the top10 poses, irrespective of their similarity/correctness, underscoring the robustness of our simple predictor. This results in an enrichment factor of 2.5 compared to a random predictor for ranking ligands within the top 25%, making it a promising approach to identify lead compounds in virtual screening.
Related collections
Most cited references30

- Record: found
- Abstract: found
- Article: found
BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology
- Record: found
- Abstract: found
- Article: not found
MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology.
- Record: found
- Abstract: found
- Article: not found