- Record: found
- Abstract: found
- Article: not found
Novel ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo
research-article
C. Korin Bullen
1 ,
Gregory M. Laird
1 ,
Christine M. Durand
1 ,
Janet D. Siliciano
1 ,
Robert F. Siliciano
1
,
2
23 March 2014

There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
HIV-1 persists in a latent reservoir (LR) despite antiretroviral therapy (ART)
1–5
. This reservoir is the major barrier to HIV-1 eradication
6,7
. Current approaches to purging the LR involve pharmacologic induction of HIV-1 transcription
and subsequent killing of infected cells by cytolytic T lymphocytes (CTL) or viral
cytopathic effects
8–10
. Agents that reverse latency without activating T cells have been identified using
in vitro models of latency. However, their effects on latently infected cells from
infected individuals remain largely unknown. Using a novel ex vivo assay, we demonstrate
that none of the latency reversing agents (LRAs) tested induced outgrowth of HIV-1
from the LR of patients on ART. Using a novel RT-qPCR assay specific for all HIV-1
mRNAs, we demonstrate that LRAs that do not cause T cell activation do not induce
significant increases in intracellular HIV-1 mRNA in patient cells; only the PKC agonist
bryostatin-1 caused substantial increases. These findings demonstrate that current
in vitro models do not fully recapitulate mechanisms governing HIV-1 latency in vivo.
Further, our data indicate that non-activating LRAs are unlikely to drive the elimination
of the LR in vivo when administered individually.
HIV-1 cure is hindered by viral persistence in a small fraction (~1/106) of resting
CD4+ T cells (rCD4s) that harbor latent but replication-competent proviruses
1–3
. Upon cellular activation, latency is reversed and replication-competent virus is
produced. Although T cell activation reverses latency, global T cell activation is
toxic, generating interest in small molecule latency-reversing agents (LRAs) that
do not activate T cells. Due to the low frequency of latently infected rCD4s in vivo,
cell models have been used to identify a number of mechanistically distinct LRAs.
These include: (1) histone deacetylase (HDAC) inhibitors, thought to function through
epigenetic and other mechanisms
11–14
; (2) disulfiram, postulated to involve nuclear factor kappa-light-chain-enhancer
of activated B cells (NF-κB)
15,16
; and (3) the bromodomain-containing protein 4 (BRD4) inhibitor JQ1, which elicits
effects through positive transcription elongation factor (P-TEFb)
17–20
. Acting through signaling pathways associated with T cell activation, protein kinase
C (PKC) agonists such as phorbol esters, prostratin
21–23
and bryostatin-1
12,24–26
also reverse latency in cell models.
Evidence that putative LRAs reverse latency ex vivo in primary rCD4s from HIV-1-infected
individuals is limited; disulfiram and the HDAC inhibitor vorinostat have been tested
in patient cells with inconsistent results
11,13,16,27,28
. Clinical trials in patients on ART are ongoing with disulfiram and the HDAC inhibitors
vorinostat, romidepsin, and panobinostat
27,29
. A recent trial of disulfiram showed no consistent evidence of latency reversal
30
. In another clinical trial, a single dose of vorinostat modestly increased intracellular
RNAs containing HIV-1 gag sequences in rCD4s of patients on ART
27
. Ex vivo treatment of patient cells with vorinostat induced outgrowth in some studies
11,13
but no virion production in another study
28
. Importantly, no LRA has been shown to reduce the size of the LR.
A consistent ex vivo validation strategy has not been employed to compare putative
LRAs. Given the costs and risks associated with clinical trials, such a strategy is
important for HIV-1 eradication research. Therefore, we utilized three independent
assays to evaluate the efficacy of LRAs in cells from HIV-1 infected individuals on
suppressive ART (participant characteristics in Supplementary Table 1).
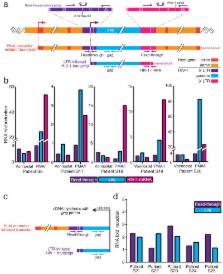
We first tested LRAs in a modified viral outgrowth assay
1
. In the original assay, patient-derived rCD4s were activated and co-cultured with
CD4+ T lymphoblasts from healthy donors to expand released virus. Induction of outgrowth
provides conclusive evidence of latency reversal. In the modified assay, T cell activation
was replaced with LRA treatment. The subsequent co-culture of patient rCD4s with healthy
donor lymphoblasts constitutes a mixed lymphocyte reaction, which induces background
reactivation of latent HIV-1
31
and complicates LRA evaluation. Therefore, we treated rCD4s with LRAs and then cultured
the cells with a transformed CD4+ T cell line (MOLT-4/CCR5) (Fig. 1a) that supports
robust HIV-1 replication but does not induce allogeneic stimulation of rCD4s (Supplementary
Fig. 1a–c). We treated five million purified rCD4s from infected individuals on ART
with single LRAs for 18 h and then co-cultured the cells with MOLT-4/CCR5 cells for
14 days to permit viral outgrowth. T cell activation with phorbol 12-myristate 13-acetate
+ ionomycin (PMA/I) served as a positive control. We concurrently measured the frequency
of latently infected cells
32
. We evaluated vorinostat, romidepsin, panobinostat, disulfiram and bryostatin-1 at
clinically relevant concentrations that effectively reversed latency in a primary
cell model (see below) and that were not toxic to rCD4s. No drug treatment induced
cell death as shown by the lack of 7-AAD staining (Fig. 1b). Surprisingly, none of
the LRAs induced viral outgrowth from cells from any individual tested while PMA/I-treated
cultures were positive for every patient with a detectable LR (Fig. 1c).
We next asked whether LRA treatment induced rapid virus release. We collected culture
supernatants from rCD4s from five infected individuals (S26–S30) after 18 h of LRA
treatment and prior to addition of MOLT-4/CCR5 cells for measurement of viral outgrowth.
PMA/I induced virus release as detected by HIV-1 mRNA in the supernatant from four
out of five individuals (S26–S29) (Fig. 1D). Bryostatin-1 treatment induced detectable
supernatant HIV-1 mRNA from one infected individual (S27), whereas no other LRA had
a measurable effect (Fig. 1d). None of the LRAs induced subsequent viral outgrowth
from these treated cells, including the cells from the single individual (S27) that
released HIV-1 mRNA after bryostatin-1 treatment (Fig. 1c).
The most widely used method to detect induction of HIV-1 transcription
16,27
in cells from infected individuals involves the measurement of RNAs containing HIV-1
gag sequences. Because this method lacks a stringent selection for poly-adenylated
RNAs, it does not exclusively detect fully elongated and correctly processed HIV-1
mRNAs. Therefore, we devised a new assay specific for intracellular HIV-1 mRNA using
a primer/probe set that detects the 3′ sequence common to all correctly terminated
HIV-1 mRNAs (Fig. 2a). We detected baseline intracellular HIV-1 mRNA in rCD4s from
ten out of 11 infected individuals. Stimulation with PMA/I for 18 h dramatically increased
intracellular HIV-1 mRNA (mean increase = 115.5-fold, Fig. 2b). However, at clinically
relevant concentrations that reverse latency in a primary cell model (Fig. 3B, C),
vorinostat, romidepsin, panobinostat, disulfiram, and JQ1 failed to increase intracellular
HIV-1 mRNA in rCD4s from infected individuals when used as single agents (Fig. 2b,
c). Bryostatin- 1 caused significant increases in some infected individuals (Fig.
2c). We observed similar results after 6 h of LRA treatment (Supplementary Fig. 2).
While no effect was seen in latently infected cells from infected individuals, LRA
treatment increased intracellular HIV-1 mRNA in a B-cell lymphoma 2 (BCL-2) transduced
primary rCD4 model of latency (Fig. 3a). LRA-induced increases in HIV-1 mRNA were
consistent with measurements of the fraction of cells that up-regulate HIV-1 gene
expression, as assessed by GFP reporter (Fig. 3b). The frequency of latent infection
in this model is substantially higher than that observed in vivo
4
. To confirm that our assay effectively detects intracellular HIV-1 mRNA increases
at frequencies of latent infection seen in vivo, we treated model cells with a known
percentage of latent infection and then serially diluted these cells into rCD4s from
uninfected individuals immediately prior to RNA isolation. We detected proportionate
increases in intracellular HIV-1 mRNA in vorinostat-treated cells down to a frequency
of 1/106 cells (Fig. 2d, e). Therefore, the lack of LRA efficacy in cells from HIV-1
infected individuals is not a result of assay insensitivity. Rather, our findings
demonstrate that freshly isolated latently infected cells from infected individuals
responded differently to LRAs than latency model cells.
RT-qPCR assays that detect gag-containing sequences in total RNA are frequently used
to detect latency reversal. These sequences do not necessarily represent bona fide
unspliced HIV-1 mRNA. HIV-1 integrates into host genes that are actively transcribed
in rCD4s
33,34
, allowing for the production of chimeric host/HIV-1 primary transcripts. Such transcripts,
initiated at host promoters, could contain gag sequence and would be indistinguishable
from LTR-initiated transcripts by conventional gag RT-qPCR assays (Fig. 4a). We therefore
designed a primer/probe set that amplifies a region of the LTR that is not transcribed
during LTR-initiated and correctly terminated HIV-1 transcription. This primer/probe
set is specific for transcripts containing read-through of the 5′ LTR or 3′ LTR, independent
of proviral orientation (Fig. 4a). We treated ten million rCD4s from infected individuals
on ART with vorinostat or PMA/I for 6 h and compared the levels of HIV-1 mRNA, read-through
transcripts, and transcripts containing gag sequence (Fig. 4a, b). We detected a small
increase (~2-fold) in transcripts containing gag sequence in vorinostat-treated rCD4s
from four out of five infected individuals, consistent with previous reports
27
(Fig. 4b). Vorinostat treatment also induced increases in read-through transcripts
(Fig. 4b) comparable to the increases in transcripts containing gag sequence but had
no effect on levels of HIV-1 mRNA (Fig. 4b).
To prove that the read-through signal is amplified from a transcript that initiated
upstream of the 5′ LTR and contains gag sequence, we primed cDNA synthesis with a
gag primer (Fig. 4c). We detected comparable, statistically significant inductions
of read-through and gag transcripts after 6 h of vorinostat treatment (Fig. 4d) (P
= 0.027, P = 0.011, respectively; ratio paired t-test of transcript copies), indicative
of read-through transcription. PMA/I induction of gag transcripts greatly exceeded
that of read-through transcripts, indicative of LTR-initiated transcription (Supplementary
Fig. 3). While not every potential LRA will induce read-through transcription by activating
a host gene, our data show that chimeric host/HIV-1 transcripts can have a confounding
effect on the RT-qPCR signal obtained with standard gag primers. Such an effect should
be taken into consideration when evaluating LRAs using conventional gag RT-qPCR assays.
The novel assays presented herein facilitated the first comparative ex vivo evaluation
of candidate LRAs. Our data demonstrate that none of the leading candidate non-T cell
activating LRAs tested significantly disrupted the LR ex vivo. The striking discordance
between the effects of non-stimulating LRAs in in vitro models of HIV-1 latency and
the ex vivo effects in rCD4s from infected individuals on ART indicates that these
models do not fully capture all mechanisms governing HIV-1 latency in vivo. These
compounds are unlikely to drive the elimination of the LR in vivo when administered
individually. The only active single agent was the PKC agonist bryostatin-1, which
is likely too toxic for clinical use. Whether other PKC agonists or other compounds
that stimulate signaling pathways associated with T cell activation can be safely
administered remains to be seen, and further progress may depend on finding safe and
active combinations of LRAs.
Methods
Cell isolation and culture
The Johns Hopkins Institutional Review Board approved this study and all research
participants in this study gave written informed consent. Infected individuals were
enrolled under the criteria of suppression of viremia to undetectable levels (<50
copies mL−1) on ART for at least 6 months. PBMC were purified using density centrifugation
from whole blood or continuous-flow centrifugation leukapheresis product. CD4+ T lymphocytes
were enriched by negative depletion (CD4+ T cell Isolation Kit, Miltenyi Biotec).
Resting CD4+ T lymphocytes were further enriched by depletion of cells expressing
CD69, CD25, or HLA-DR (CD69 MicroBead Kit II, Miltenyi Biotec; CD25 MicroBeads, Miltenyi
Biotec; Anti-HLA-DR MicroBeads, Miltenyi Biotec). Purity of resting CD4+ lymphocytes
was verified by flow cytometry and was typically greater than 95%. With the exception
of experiments designed to detect viral outgrowth, cells were cultured with 10 μM
T20 to prevent new infection events.
Treatment of rCD4s with LRAs
rCD4s were treated with the following concentrations: 335 nM vorinostat, 40 nM romidepsin,
30 nM panobinostat, 500 nM disulfiram, 1 μM JQ1, 10 nM bryostatin-1, or 50 ng mL−1
PMA plus 1 μM ionomycin.
MOLT-4/CCR5 outgrowth assay
Five million purified rCD4s were treated with LRA for 18 h in a volume of 1 mL RPMI
+ 10% FBS. Cells were then resuspended, transferred to a microcentrifuge tube and
pelleted. Cells were washed with 1 mL sterile PBS to remove residual drug and pelleted.
rCD4s were then cultured with MOLT-4/CCR5 cells in 8 mL RPMI + 10% FBS in individual
wells in 6 well plates. After 4 days of culture, cells were resuspended and split
into two wells of a 6 well plate with the media volume adjusted to 8 mL per well.
After 7 days of culture, wells were resuspended and split 1:2 with the media volume
adjusted to 8 mL per well. Viral outgrowth was assessed at 14 days using the Alliance
HIV-1 p24 antigen ELISA kit (Perkin Elmer).
Cell lines
MOLT-4/CCR5 cells from Dr. Masanori Baba, Dr. Hiroshi Miyake, and Dr. Yuji Iizawa
were obtained from the NIH AIDS Reagent Program, NIAID, NIH
35
.
Generation of latently HIV-1 infected BCL-2 transduced cells
Latently HIV-1 infected BCL-2 transduced cells were generated as described previously
36
. Briefly, primary CD4+ lymphoblasts were transduced with BCL-2 and allowed to return
to a resting state in the absence of exogenous cytokines. BCL-2 transduced cells were
then activated and expanded in the presence of exogenous IL-2. After expansion, cells
were activated again and infected with a recombinant HIV-1 containing GFP in place
of the env gene. After infection, cells were allowed to return to a resting state
and GFP-negative cells were isolated via cell sorting. This population of cells includes
the fraction of cells that are in vitro latently infected. Reversal of latency is
assessed by flow cytometry analysis of GFP expression.
Measurement of intracellular HIV-1 RNA transcripts
Cells were treated with each LRA in triplicate in the presence of 10 μM T20 (5 × 106
cells for experiments measuring only HIV mRNA and 10 × 106 cells for experiments measuring
multiple transcripts). Cells were pelleted in RNase-free low binding microcentrifuge
tubes and subsequently lysed with 1 mL of TRIzol Reagent (Invitrogen). RNA was isolated
using the manufacturer’s protocol. For experiments in which multiple transcripts were
measured, a DNase digest was performed using TURBO DNase (Ambion). RNA was subsequently
re-extracted using Acid-Phenol:Choloroform, pH 4.5 (Ambion) per manufacturer’s protocol.
cDNA synthesis was performed using qScript cDNA Supermix containing random hexamers
and oligo-dT primers(Quanta Biosciences). Gag specific cDNA synthesis was performed
using Superscript III First-Strand Synthesis (Invitrogen) using only a gag primer
(sequence listed below). A fraction of the RNA was retained for RT(−) control reactions.
Real-time PCR was performed in triplicate using TaqMan® Universal PCR Master Mix (Applied
Biosystems) on an ABI7900 Real-Time PCR machine. Approximately one million cell equivalents
of cDNA or RNA (for no-RT control reactions) template was used in each PCR reaction.
Primers and probes are listed below. The cycling parameters were as follows: (i) 2
min at 50°C; (ii) 10 min at 95°C; and (iii) 45–50 cycles at 95°C for 15 and then 60°C
for 60 s. Molecular standard curves were generated using serial dilutions of a TOPO
plasmid containing the 5′ LTR, Gag, or the last 352 nucleotides of viral genomic RNA
plus 30 deoxyadenosines.
Results from the triplicate samples for each drug treatment were averaged and presented
as fold change relative to DMSO control (mean ± s.e.m.) or copies of HIV-1 mRNA per
million rCD4 equivalents. The limit of quantification was set as the dilution point
at which the Ct of the plasmid molecular standard replicates had a standard deviation
> 0.5. We determined that the limit of quantification for all transcripts was 10 copies.
A PCR signal of less than 10 copies (1–9 copies) was treated as 10 copies in calculations
of fold change and marked as 10 copies on graphs depicting RNA copies. Undetectable
PCR signal was treated as 10 copies in calculations of fold change and marked as 1
copy on graphs depicting RNA copies. Levels of RNA polymerase II (Pol2) and Glucose-6-phosphate
dehydrogenase (G6PD) RNA were also measured for each sample to use as an endogenous
control. Voronistat, romidepsin, panobinostat, JQ1 and PMA/I treatment consistently
increased expression Pol2 and G6PD. Samples treated with the same drug had even levels
of Pol2 and G6PD, indicating that the template inputs were approximately equal.
Measurement of supernatant HIV-1 mRNA
HIV-1 mRNA was extracted from 0.2mL of supernatant from five million cultured rCD4s
after 18 h of LRA treatment using the ZR-96 Viral RNA kit (Zymo Research). cDNA synthesis
was performed using qScript cDNA Supermix (Quanta Biosciences). Real-time PCR was
performed using TaqMan Fast Advanced mastermix (Applied Biosystems) on an ABI Viia
7 Real-Time PCR machine. Primers and probes listed below. Manufacturer’s thermal cycling
conditions were used. Molecular standard curve was generated as described above.
Primer and probe sequences
Nucleotide coordinates are indicated relative to HXB2 consensus sequence.
HIV-1 mRNAs were detected using the following primers and probe, modified from Shan
et al.
37
:
Forward (5′→3′) CAGATGCTGCATATAAGCAGCTG (9501–9523)
Reverse (5′→3′) TTTTTTTTTTTTTTTTTTTTTTTTGAAGCAC (9629-poly A)
Probe (5′→3′) FAM-CCTGTACTGGGTCTCTCTGG-MGB (9531–9550)
Transcripts containing HIV-1 gag sequence were detected using the following primers
and probe, described previously
27
.
Forward (5′→3′) ACATCAAGCAGCCATGCAAAT (1368–1388)
Reverse (5′→3′) TCTGGCCTGGTGCAATAGG (1453–1471)
Probe (5′→3′) VIC-CTATCCCATTCTGCAGCTTCCTCATTGATG-TAMRA (1401–1430)
Chimeric host/HIV-1 read-through transcripts were detected using the following primers
and probe:
Forward (5′→3′) CAGATGCTGCATATAAGCAGCTG (416–438, 9501–9523)
Reverse (5′→3′) CACAACAGACGGGCACACAC (556–575, 9641–9660)
Probe (5′→3′) FAM-CCTGTACTGGGTCTCTCTGG-MGB (446–465, 9531–9550)
cDNA synthesis reaction with gag primer sequence:
Reverse (5′→3′) GTCACTTCCCCTTGG (1480–1494)
Supplementary Material
1
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy.
J. Gallant, R Brookmeyer, J Margolick … (1998)
- Record: found
- Abstract: found
- Article: not found
Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy.
T Chun, L Stuyver, S. B. Mizell … (1997)

- Record: found
- Abstract: found
- Article: found
Rapid Quantification of the Latent Reservoir for HIV-1 Using a Viral Outgrowth Assay
Gregory M. Laird, Evelyn Eisele, S. Alireza Rabi … (2013)