- Record: found
- Abstract: found
- Article: found
Human malarial disease: a consequence of inflammatory cytokine release
Read this article at
Abstract
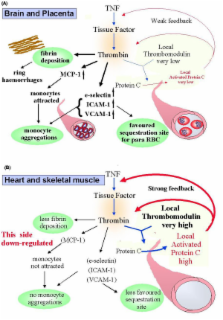
Malaria causes an acute systemic human disease that bears many similarities, both clinically and mechanistically, to those caused by bacteria, rickettsia, and viruses. Over the past few decades, a literature has emerged that argues for most of the pathology seen in all of these infectious diseases being explained by activation of the inflammatory system, with the balance between the pro and anti-inflammatory cytokines being tipped towards the onset of systemic inflammation. Although not often expressed in energy terms, there is, when reduced to biochemical essentials, wide agreement that infection with falciparum malaria is often fatal because mitochondria are unable to generate enough ATP to maintain normal cellular function. Most, however, would contend that this largely occurs because sequestered parasitized red cells prevent sufficient oxygen getting to where it is needed. This review considers the evidence that an equally or more important way ATP deficency arises in malaria, as well as these other infectious diseases, is an inability of mitochondria, through the effects of inflammatory cytokines on their function, to utilise available oxygen. This activity of these cytokines, plus their capacity to control the pathways through which oxygen supply to mitochondria are restricted (particularly through directing sequestration and driving anaemia), combine to make falciparum malaria primarily an inflammatory cytokine-driven disease.
Related collections
Most cited references353
- Record: found
- Abstract: found
- Article: not found
HMG-1 as a late mediator of endotoxin lethality in mice.
- Record: found
- Abstract: not found
- Article: not found
Avian influenza A (H5N1) infection in humans.
- Record: found
- Abstract: found
- Article: not found