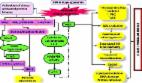
Introduction Diabetes Mellitus (DM) is a chronic disorder characterized by impaired metabolism of glucose and lipids due to defect in insulin secretion (beta cell dysfunction) or action (insulin resistance). The characteristic properties of diabetes mellitus are chronic hyperglycemia, microvascular (eg. retina, renal glomerulus and peripheral nerve) as well as macrovascular (eg. atherosclerosis, coronary artery disease (CAD), stroke) pathologies with more than 17.5 million deaths worldwide attributable to cardiovascular complications [1]. According to International Diabetes Federation (IDF) Diabetes Atlas 5th Edition-2012 update, 371 million people have been reported with DM and the number is expected to rise to >552 million by 2030. The 2012 Indian statistics showed 63.0 million diabetic cases and a prevalence of 8.37% in adult population [2] while a 4.0% prevalence of type 2 diabetes mellitus (T2DM) was reported in North Indian population [3]. The currently favored hypothesis is oxidative stress leading to insulin resistance (IR), β-cell dysfunction, impaired glucose tolerance (IGT) and ultimately T2DM. Furthermore, oxidative stress has been implicated as the underlying cause of both macrovascular and microvascular complications associated with T2DM. It is believed that therapies aimed at reducing oxidative stress would benefit patients with T2DM and also those at risk. The accumulation of glucose and fatty acids within muscles, adipose tissue and pancreatic cells combined with sedentary lifestyle lead to the generation of excess reactive metabolites (RMs). Oxidative stress and RMs are interrelated terms defined in general as excess formation and/or insufficient removal of highly reactive molecules such as reactive oxygen species (ROS), reactive nitrogen species (RNS) and reactive thiyl species (RTR). This review aims to explain the role of such RMs and association of antioxidant gene polymorphisms with T2DM. Types of reactive metabolites (RMs) ROS: oxygen derived free radicals (ODFR) and oxygen derived non radicals (ODNR) Oxygen derived free radical and non-radical reactive species are generated in metabolic pathways of biological systems. ODFR include superoxide ( O 2 − ), hydroxyl (•OH), peroxyl (•RO2), hydroperoxyl ( H RO 2 − ) while ODNR include hydrogen peroxide (H2O2) and hydrochlorous acid (HOCl). These metabolites are responsible for lipid and protein modifications in case of oxidative stress [4]. Basal oxidative cellular metabolism generates a number of oxygen-derived free radical species through the activation of enzymes that produce superoxide anions and/or byproducts of mitochondrial respiration [5]. Reactive nitrogen species (RNS): nitrogen derived free radicals (NDFR) and nitrogen derived non radicals (NDNR) Like ROS, RNS can be classified into radical and non-radical species. NDFR include nitric oxide (•NO), nitrogen dioxide ( N O 2 − ) while NDNR include alkyl peroxynitrates (RONOO−), nitrous oxide (HNO2) and peroxynitrite (ONOO−). O 2 − , •NO and ONOO− are the most widely studied species and play important roles in cardiovascular complications [5]. Nitric oxide (•NO) is responsible for formation of many end-products involved in oxidative stress directly or indirectly after reaction with oxygen. •NO-derived RNS react with aromatic amino acids, lipids and thiols resulting in lipid and protein modifications [4]. Leukocyte peroxidases are involved in the formation of •NO2 after utilization of H2O2 and NO 2 − as substrates. •NO2, •OH and ONOOH are responsible for damages related to oxidative stress eg. oxidation, nitrosation and nitration reactions [5,6]. Reactive thiyl and tyrosyl radicals (RTR) RMs play a major role in generation of thiyl radicals (RS•) and their derivatives, sulfinyl (RSO•) and disulfide anion radical (RSSR•−) [7]. ROS and RNS induce protein S-glutathionylation either by protein thiol oxidative/nitrosative modification or by presence of protein thiyl radical (R–S•), sulfenic acid (R–SOH) and S-nitrosothiol (R–SNO) [7]. Thiyl radicals (TR) may be formed by •OH, ONOO− and/or Fe3+ mediated oxidation of thiols. TR may also be derived from sulfur containing moieties including disulfide, thioester or thioether functionalities under conditions of oxidative stress. Once formed, TR not only reacts with themselves and oxygen but also oxidize biological electron donors including ascorbic acid, NADH and ferricytochrome C. Myeloperoxidase uses H2O2 generated by the cells to oxidize l-tyrosine to tyrosyl radical. RTR radical initiates lipid peroxidation which may be of pivotal importance in transforming low density lipoprotein (LDL) into atherogenic particles [8]. Glutathione (GSH) is a tripeptide, γ-glutamyl-l-cysteinylglycine detected in all mammalian tissues and is present in disulfide form of glutathione (GSSG) [9]. Thiol status/redox balance determined by redox pair 2GSH/GSSG is an indicator of redox homeostasis or oxidative stress inside the cell. S-glutathionylation has been proposed as a posttranslational modification which is able to protect proteins from over-oxidizing environments in a wide range of diseases including diabetes mellitus [9]. Production of reactive metabolites (RMs) Production of RMs mainly superoxides ( O 2 − ) have been found in a variety of predominating cellular enzyme systems including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase (XO), cyclooxygenase (COX), uncoupled endothelial nitric oxide synthase (eNOS) and myeloperoxidase (MPO) [10] (Table 1). The various sources of ROS and action of antioxidant enzymes have been represented in Fig. 1. NADPH oxidase uses NADPH as a substrate and is considered as an important source of ROS generation in vascular cells [4]. The lipoxygenase (LPO) and COX generate ROS indirectly by promoting formation of inflammatory mediators. RM production may result from action of arachidonic acid metabolizing enzymes including cytochrome P-450, LPO, COX and those in the mitochondrial respiratory chain [11]. Arachidonic acid (AA) is cleaved from the membrane by phospholipase A2 (PLA2) and is then metabolized by 5-LPO in the presence of its accessory protein 5-lipoxygenase activating protein (FLAP) to form leukotrienes (LTs) [12]. AA is also metabolized by COX to form members of another family of inflammatory mediators, the prostaglandins (PGs) [12]. Mitochondria also generate superoxides as electrons are transferred from complexes I to IV during normal cellular respiration. XO, which converts hypoxanthine and xanthine to uric acid, is an additional source of ROS [12]. Finally, eNOS uncouples to generate superoxide in preference to NO [13]. Reactive metabolites and Type 2 diabetes mellitus There are four main molecular mechanisms implicated in glucose-mediated vascular damage viz. increased polyol pathway flux; increased production of advanced glycation end-product (AGE); activation of protein kinase C (PKC) isoforms, sorbitol, cytokines and prostanoids along with increased hexosamine pathway flux [16,17] (Fig. 2). O 2 − leads to several damaging pathways resulting in micro and macrovascular complications in diabetes which accelerates the formation of advanced glycation end products (AGE), polyol pathway and phospholipase C (PLC) [16,17]. O 2 − and H2O2 stimulate stress-related signaling mechanisms such as nuclear factor kappa light chain enhancer of activated B cells (NF-κB), mitogen activated protein kinase (p38-MAPK) and janus kinase signal transducer and activator of transcription (JAK-STAT) resulting in vascular smooth muscle cell (VSMC) migration and proliferation [16]. H2O2 also mediates apoptosis and pathological angiogenesis in endothelial cells [6]. Pathway-selective insulin resistance also results in decreased endothelial production of the anti-atherogenic molecule, nitric oxide [17]. Additional stress-sensitive kinases that are reported to be involved in insulin receptor substrate (IRS)-mediated insulin resistance include several isozymes of protein kinase C (PKC) such as PKCβ, PKCγ and inhibitor kinase beta NF-κB (IKKβ-NFκB) [16,18]. Once activated, these kinases are able to phosphorylate multiple targets including insulin receptors and IRS proteins such as IRS-1 and IRS-2. Oxidative stress activates serine kinases by various molecular mechanisms resulting in phosphorylation of serine in IRS, which induces insulin resistance [19,20]. This reflects decreased activity of vasodilators such as nitric oxide, increased activity of vasoconstrictors such as angiotensin II and endothelin-1 and elaboration of permeability factors such as vascular endothelial growth factor (VEGF) [16,21]. Quantitative and qualitative abnormalities of extracellular matrix contribute to an irreversible increase in vascular permeability. Microvascular cell loss occurs with time as a result of programmed cell death and progressive capillary occlusion. Both occur due to extracellular matrix and overproduction induced by growth factors such as transforming growth factor-β (TGF-β) and deposition of plasma proteins [13,16,21]. A causative link among hyperglycemia, mitochondrial ROS generation, oxidative stress and development of complications has been suggested which plays a key role in the pathogenesis of diabetes [4,22]. Damage due to RMs is associated with complex metabolic and structural changes in the body for example, oxidation of low-density lipoproteins (Ox-LDL) which are taken up by scavenger receptors in macrophages leading to foam cell formation and atherosclerotic plaques [4,23]. ROS-induced peroxidation of membrane lipids alters the structure and fluidity of biological membranes which ultimately affects cellular function [24]. In T2DM, such activation of stress-sensitive pathways and elevations in glucose and free fatty acid (FFA) levels lead to both insulin resistance and impaired insulin secretion. In diabetic arteries, endothelial dysfunction is a result of both insulin resistance and hyperglycemia [21]. Diabetic dyslipidaemia is a result of insulin resistance and hyperglycemia [21]. Hyperglycemia seems to cause reduced expression of heparan sulfate proteoglycan and perlecan on hepatocytes resulting in raised levels of atherogenic cholesterol-enriched apolipoprotein B-containing remnant particles [21]. Postprandial hyperglycemia may be more predictive of atherosclerosis than fasting plasma glucose level or hemoglobin A1c (HbA1c) [16,21]. Changes defined lead to edema, ischemia and hypoxia-induced neovascularization in the retina, proteinuria, mesanglial matrix expansion and glomerulosclerosis in the kidney and multifocal axonal degeneration in peripheral nerves [21]. Diabetes related microvascular pathology is the leading cause of blindness, end stage renal disease and a variety of debilitating neuropathies [21]. The pathogenesis of atherosclerosis also begins with endothelial dysfunction. Atherosclerotic macrovascular disease affects arteries that supply the heart, brain and lower extremities. As a result, patients with diabetes have a much higher risk of myocardial infarction, stroke and limb amputation [22]. In healthy individuals both enzymatic and non-enzymatic antioxidant defense play important roles in scavenging ROS and RNS (Table 2). Impaired antioxidant defense increases oxidative stress and contributes to the development of T2DM and diabetic cardiovascular disease (CVD). Our recent studies have shown that activity levels of SOD and GST were significantly lower in T2DM patients than in healthy subjects [25]. O 2 − produces hydrogen peroxide (H2O2) on its dismutation by copper superoxide dismutase (Cu-SOD) and manganese-superoxide dismutase (Mn-SOD). Hydrogen peroxide (H2O2) produces hydroxyl radical (•OH) by reaction with reduced transition metals (Fe or Cu) i.e. fenton reaction and can be metabolized to HOCl by MPO [10]. H2O2 is converted to H2O and O2 by glutathione peroxidase (GSH-Px) or catalase (CAT) in the mitochondria and lysosomes respectively. The inner mitochondrial membrane also contains vitamin E which is a powerful antioxidant as it can accept unpaired electrons to produce a stable product [22]. Antioxidant genes and polymorphisms Superoxide dismutase (SOD) Once formed, O 2 − is dismutated enzymatically to H2O2 and oxygen by the SOD family of antioxidant enzymes which include intracellular (Cu Zn-SOD), mitochondrial (Mn-SOD), and extracellular (EC-SOD) enzymes also referred to as SOD types 1, 2 and 3 respectively [13]. An increase in Cu Zn-SOD (SOD1) expression projects human smooth muscle cells against oxidative injury. Ox-LDL causes an increase in the DNA binding activity of activator protein-1 and NF-κB which is inhibited by Cu Zn-SOD overexpression. Cu Zn-SOD (SOD1) gene is located on chromosome 4p15.1–15.3 while Mn-SOD (SOD2) gene is located on chromosome 6q25. A functional polymorphism in exon 2 of SOD2 gene A16V (C/T) (rs4880) was identified that resulted in structural alterations in the mitochondrial targeting domain, implicating its decreased antioxidant potential to limited post-transcriptional transport. The substitution from valine to alanine was shown to induce a 30–40% increase in Mn-SOD activity in mitochondria resulting in reduced risk of coronary artery disease (CAD) and acute myocardial infarction (AMI) [26]. Individuals harboring the valine variant had an increased carotid intima-to-media thickness (IMT) and were at increased risk for CAD and AMI [27]. Increased oxidative stress is the leading cause of post-translational covalent modifications in SOD e.g. nitration, phosphorylation, glutathionylaion, and glycation which results in decreased enzyme activity [28]. Polymorphic conditions of SOD1 35 A/C (rs2234694) and SOD2 A16V (C/T) showed that serum SOD activity was higher in individuals with ‘CC’ genotype than ‘TT’ genotype of SOD2 gene and higher in ‘AA’ as compared to ‘CC’ genotype of SOD1 gene. Better diabetes control was found in patients with ‘CC’ genotype of SOD2 gene. Significantly different allele and genotype frequencies of SOD2 gene polymorphism were found among T2DM patients with and without macroangiopathy, diabetic retinopathy in Chinese, diabetic macular edema (DME) and albuminuria in Koreans [29–31]. EC-SOD (SOD3) is bound to matrix and EC proteoglycans. EC-SOD (SOD3) gene is located on chromosome 21q22.11. Clinical studies have shown a decrease in EC-SOD activity in aged, African–Americans with hypertension, patients with vasospastic angina, thoracic aortic aneurysm and calcified aortic stenosis. Serum SOD activity was significantly decreased in T2DM subjects compared to control subjects [32,33]. The variant R213G in the heparin-binding domain of EC-SOD has been linked to cardiovascular disease risk. This polymorphism was linked to increased risk of ischemic heart disease in a Danish case control study as well [34]. Catalase Catalase (CAT) is present in peroxisomes and exists as a dumbell-shaped tetramer of four identical subunits. It rapidly catalyzes the decomposition of hydrogen peroxide into less reactive oxygen and water molecules. CAT deficiency was known to lead to the development of T2DM [35,36]. CAT gene is located on chromosome 11p13. Exon 2 and neighboring introns of the CAT gene were thought to be mutation hot spots for T2DM susceptibility [37,38]. Under conditions of oxidative stress, modification of cystein to cysteic acid leads to tyrosyl nitration of catalase and decreased activity [39,40]. The exon 9-262C/T polymorphism in CAT gene was examined in types 1, 2 and gestational diabetes (GD) and complications such as diabetic retinopathy (DR), diabetic nephropathy (DN), ischemic heart disease (IHD) and CVD [41]. This functional polymorphism also contributed to the development of T2DM and its complications [42,43]. ‘AT’ genotype of −21A/T polymorphism of CAT gene may increase the risk of T2DM in north Indians [44]. Glutathione peroxidases Glutathione peroxidases (GPx) are selenocysteine-containing enzymes that catalyze the reduction of H2O2 and lipid hydroperoxides to H2O and lipid alcohols respectively in a reaction that utilizes glutathione (GSH) as a reducing co-substrate [41]. GPx may also function as ONOO− reductase. There are 5 known forms of GPx: cellular (GPx-1), gastrointestinal (GPx-2), plasma (GPx-3), phospholipid (GPx-4), and sperm (snGPx). The importance of GPx family of antioxidant enzymes limits the oxidative risk for atherothrombosis. GPx-1 is an ubiquitous antioxidant enzyme whose deficiency has been shown to promote endothelial dysfunction, heart failure and abnormal structural changes in vasculature and myocardium [49]. Interestingly, hyperhomocystinemia appears to enhance vascular oxidative stress and atherothrombosis, in part by suppressing expression of the GPx-1 gene which is located on chromosome 3p21.3. Erythrocyte GPx-1 activity and association of GPx-1 genotypes was shown as independent determinants of cardiovascular risk and CAD [50]. A polyalanine sequence polymorphism in exon 1 of GPx-1 gene produces 3 alleles with 5, 6, or 7 alanine repeats. Men with at least one 6-alanine repeat had significantly increased risk of CAD. The Pro198Leu C/T polymorphism in GPx-1 gene increased carotid intima-media thickness (IMT), prevalence of cardiovascular and peripheral vascular disease in Japanese patients with T2DM [41,51]. Out of a large number of factors mediating atherosclerotic risk in plasma, focus lays on GPx-3, the essential extracellular peroxidase and its role in modulating oxidative stress. Deficiency of GPx-3 was associated with decreased nitric oxide bioavailability and increased platelet dependent thrombosis. There was a reduction in plasma GPx-3 activity with increased platelet activation and cerebrovascular arterial thrombosis [52,53]. GPx-3 promoter revealed seven polymorphisms that are tightly linked and form two novel haplotypes, out of which one was associated with hypoxic conditions, arterial thrombotic stroke and cerebral venous thrombosis [45]. Over expression of GPx-4 reduces oxidized phospholipids, cholesterol hydroperoxides as well as proinflammatory lipid peroxides generated by LPO and COX thereby decreasing vascular oxidative stress and progression of atherosclerosis [54]. Glutathione-S-transferases The glutathione-S-transferases (GSTs) are dimeric cytosolic xenobiotic-metabolizing enzymes that catalyze the conjugation of an active xenobiotic to GSH, an endogenous water-soluble substrate and detoxify reactive electrophiles such as those contained in tobacco smoke. In addition to their catalytic role in detoxification, GSTs were also found to possess selenium-independent peroxidase activity with hydroperoxides, steroid isomerization capacity, binding and transport of bilirubin, heme, bile salts and steroids in a process that is associated with a decrease in enzymatic activity [55]. Several studies have found an association between GST polymorphisms and decreased enzymatic activity and atherosclerosis. Elevated levels of plaque DNA damage as well as levels of inflammatory markers such as C-reactive protein (CRP), fibrinogen and adhesion molecules were detected in individuals with GST M1 ⁎ 0 null allele. GST M1, T1 and P1 have been reported to be involved in T2DM development and various diabetes related complications [48,56–59]. Our present study indicates that individually Val105Val of GSTP1 gene and multiple combinations GST isoforms increase the risk of having T2DM in north Indian population [44]. Microsomal GST3 encoded by MGST3 gene, which maps to chromosome 1q23 is a potential susceptibility gene linked to T2DM in Pima Indians, Caucasian and Chinese populations [60]. Nitric oxide synthase Nitric oxide (NO) plays a fundamental role in the regulation of endothelial function and vascular tone in many organs including kidney. It inhibits platelet aggregation, leukocyte adhesion to vascular endothelium and oxidation of low-density lipoprotein (LDL) [46]. Upon release, NO diffuses rapidly through the cell membrane and relaxes neighboring vascular smooth cells through the production of cyclic guanine 3′5′-monophosphate (cGMP). cGMP then activates the protein kinase G family, leading to a cascade of responses at the levels of transcription and translation. Inducible nitric oxide synthase (NOSI/iNOS), neural NOS (NOS II/nNOS) and endothelial NOS (NOSIII/eNOS) are the three isoforms of NOS. Clinically, eNOS uncoupling has been associated with hypertension, diabetes mellitus, hypercholesterolemia and atherosclerosis [61]. Impairment of NO production causes endothelial dysfunction which contributes to the development of insulin resistance, T2DM, chronic renal failure and cardiovascular complications including hypertension and hypercholesterolemia [62]. eNOS or NOSIII gene, mapped to chromosome 7q36 is highly polymorphic and several studies have been undertaken to investigate the potential association of polymorphisms and risk of atherothrombotic vascular disease in Caucasian and Asian populations. SNPs in the promoter region (−786 T/C), G/T substitution at nucleotide 894 in exon 7 leading to an amino acid change (Glu298Asp) and a 27 bp variable number of tandem repeats (27 bp-VNTR) in intron 4 have received much attention because of their functional relevance to eNOS activity and association with cardiovascular and renal diseases [46]. The Glu298Asp gene polymorphism is responsible for a decrease in basal NO production and increased frequency of hypertension [63]. Additionally, eNOS Glu298Asp can interact with gene polymorphisms of other endogenous antioxidant enzymes in T2DM patients [14,15,62,64]. In atherosclerosis, vascular smooth muscle cells, monocytes, macrophages, and dendritic cells all express iNOS. Induction of iNOS may occur following exposure to inflammatory cytokines, including interleukin-1β (IL-1 β), interferon-γ (INF-γ), and tumor necrosis factor-α (TNF-α). In contrast to eNOS, iNOS binds Ca2+/calmodulin tightly and does not require an increase in intracellular Ca2+ for activation [65]. The presence of iNOS localized to macrophages and vascular smooth muscle cells was found to colocalize with oxidized lipid and protein derivatives found in atherosclerotic plaques [66]. No association of eNOS Glu298Asp (rs1799983), eNOS 4a/b and iNOS Ser608Leu (rs2297518) polymorphisms was found in T2DM patients with diabetic nephropathy [47]. It has therefore been widely used as a biomarker for oxidative stress and T2DM. Conclusions T2DM and oxidative stress have a clinical and genetic correlation which needs to be established to a greater extent. The overall play of the RMs has shown to be the lead cause of late onset insulin resistance. These RMs are generated inside the body of normal individuals in a scheduled manner and are in feedback control with the antioxidant system. Glycemic load and RMs are highly interrelated leading to various harmful molecular mechanisms implicated in glucose-mediated vascular damage such as increased polyol pathway flux; increased production of advanced glycation end-product (AGE); activation of protein kinase C (PKC) isoforms, sorbitol, cytokines and prostanoids along with increased hexosamine pathway flux. These pathways lead to increased expression of other signaling pathways which result in insulin resistance and T2DM. This review was undertaken to understand the role of RMs in T2DM and provides a lead for future research in identifying antioxidant gene variants and risk genotypes in populations of different ethnicity (Table 3). Funding The authors have received support from Indian Council of Medical Research (ICMR), New Delhi (Grant number 5/3/8/74/2009-RHN, 2011).