- Record: found
- Abstract: found
- Article: found
Autosomal dominant polycystic kidney disease: updated perspectives
Abstract
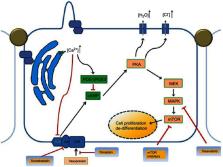
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited multisystem disorder, characterized by renal and extra-renal fluid-filled cyst formation and increased kidney volume that eventually leads to end-stage renal disease. ADPKD is considered the fourth leading cause of end-stage renal disease in the United States and globally. Care of patients with ADPKD was, for a long time, limited to supportive lifestyle measures, due to the lack of therapeutic strategies targeting the main pathways involved in the pathophysiology of ADPKD. As the first FDA approved treatment of ADPKD, Vasopressin (V 2) receptor blocking agent, tolvaptan, is an urgently awaited advance for ADPKD patients. In our review, we also shed some lights on what is beyond Tolvaptan as there are other medications in the pipeline and many medications have been or are currently being studied in clinical trials such as Tesevatinib, Metformin and Pravastatin, with the goal of slowing the rate of progression of ADPKD by reducing the increase in total kidney volume or maintaining eGFR. Here, we review updates in the perspectives and management of ADPKD.
Most cited references84
- Record: found
- Abstract: found
- Article: not found
Autosomal dominant polycystic kidney disease.
- Record: found
- Abstract: found
- Article: not found
Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease
- Record: found
- Abstract: found
- Article: not found