- Record: found
- Abstract: found
- Article: found
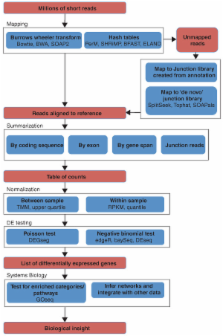
From RNA-seq reads to differential expression results
review-article
22 December 2010
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Many methods and tools are available for preprocessing high-throughput RNA sequencing data and detecting differential expression.
Related collections
Most cited references49
- Record: found
- Abstract: found
- Article: not found
Gene Ontology: tool for the unification of biology
Michael Ashburner, Catherine A. Ball, Judith Blake … (2002)
- Record: found
- Abstract: found
- Article: not found
Small-sample estimation of negative binomial dispersion, with applications to SAGE data.
Mark Robinson, Gordon K. Smyth (2008)
- Record: found
- Abstract: found
- Article: not found
De novo assembly and analysis of RNA-seq data.
Gordon Robertson, Jacqueline Schein, Readman Chiu … (2010)