- Record: found
- Abstract: found
- Article: found
3Drefine: an interactive web server for efficient protein structure refinement
Read this article at
Abstract
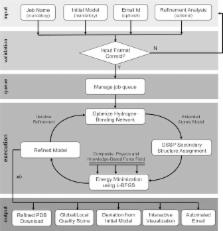
3Drefine is an interactive web server for consistent and computationally efficient protein structure refinement with the capability to perform web-based statistical and visual analysis. The 3Drefine refinement protocol utilizes iterative optimization of hydrogen bonding network combined with atomic-level energy minimization on the optimized model using a composite physics and knowledge-based force fields for efficient protein structure refinement. The method has been extensively evaluated on blind CASP experiments as well as on large-scale and diverse benchmark datasets and exhibits consistent improvement over the initial structure in both global and local structural quality measures. The 3Drefine web server allows for convenient protein structure refinement through a text or file input submission, email notification, provided example submission and is freely available without any registration requirement. The server also provides comprehensive analysis of submissions through various energy and statistical feedback and interactive visualization of multiple refined models through the JSmol applet that is equipped with numerous protein model analysis tools. The web server has been extensively tested and used by many users. As a result, the 3Drefine web server conveniently provides a useful tool easily accessible to the community. The 3Drefine web server has been made publicly available at the URL: http://sysbio.rnet.missouri.edu/3Drefine/.
Related collections
Most cited references17
- Record: found
- Abstract: not found
- Article: not found
A solution for the best rotation to relate two sets of vectors
- Record: found
- Abstract: found
- Article: not found
