- Record: found
- Abstract: found
- Article: found
The influence of a priori grouping on inference of genetic clusters: simulation study and literature review of the DAPC method
Read this article at
Abstract
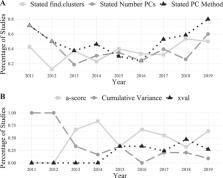
Inference of genetic clusters is a key aim of population genetics, sparking development of numerous analytical methods. Within these, there is a conceptual divide between finding de novo structure versus assessment of a priori groups. Recently developed, Discriminant Analysis of Principal Components (DAPC), combines discriminant analysis (DA) with principal component (PC) analysis. When applying DAPC, the groups used in the DA (specified a priori or described de novo) need to be carefully assessed. While DAPC has rapidly become a core technique, the sensitivity of the method to misspecification of groups and how it is being empirically applied, are unknown. To address this, we conducted a simulation study examining the influence of a priori versus de novo group designations, and a literature review of how DAPC is being applied. We found that with a priori groupings, distance between genetic clusters reflected underlying F ST. However, when migration rates were high and groups were described de novo there was considerable inaccuracy, both in terms of the number of genetic clusters suggested and placement of individuals into those clusters. Nearly all (90.1%) of 224 studies surveyed used DAPC to find de novo clusters, and for the majority (62.5%) the stated goal matched the results. However, most studies (52.3%) omit key run parameters, preventing repeatability and transparency. Therefore, we present recommendations for standard reporting of parameters used in DAPC analyses. The influence of groupings in genetic clustering is not unique to DAPC, and researchers need to consider their goal and which methods will be most appropriate.
Related collections
Most cited references50
- Record: found
- Abstract: found
- Article: not found
Detecting the number of clusters of individuals using the software structure: a simulation study
- Record: found
- Abstract: found
- Article: not found
Fast model-based estimation of ancestry in unrelated individuals.
- Record: found
- Abstract: found
- Article: not found