- Record: found
- Abstract: found
- Article: found
Global analyses of Ceratocystis cacaofunesta mitochondria: from genome to proteome
Read this article at
Abstract
Background
The ascomycete fungus Ceratocystis cacaofunesta is the causal agent of wilt disease in cacao, which results in significant economic losses in the affected producing areas. Despite the economic importance of the Ceratocystis complex of species, no genomic data are available for any of its members. Given that mitochondria play important roles in fungal virulence and the susceptibility/resistance of fungi to fungicides, we performed the first functional analysis of this organelle in Ceratocystis using integrated “omics” approaches.
Results
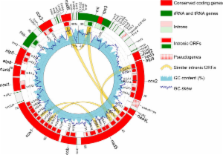
The C. cacaofunesta mitochondrial genome (mtDNA) consists of a single, 103,147-bp circular molecule, making this the second largest mtDNA among the Sordariomycetes. Bioinformatics analysis revealed the presence of 15 conserved genes and 37 intronic open reading frames in C. cacaofunesta mtDNA. Here, we predicted the mitochondrial proteome (mtProt) of C. cacaofunesta, which is comprised of 1,124 polypeptides - 52 proteins that are mitochondrially encoded and 1,072 that are nuclearly encoded. Transcriptome analysis revealed 33 probable novel genes. Comparisons among the Gene Ontology results of the predicted mtProt of C. cacaofunesta, Neurospora crassa and Saccharomyces cerevisiae revealed no significant differences. Moreover, C. cacaofunesta mitochondria were isolated, and the mtProt was subjected to mass spectrometric analysis. The experimental proteome validated 27% of the predicted mtProt. Our results confirmed the existence of 110 hypothetical proteins and 7 novel proteins of which 83 and 1, respectively, had putative mitochondrial localization.
Conclusions
The present study provides the first partial genomic analysis of a species of the Ceratocystis genus and the first predicted mitochondrial protein inventory of a phytopathogenic fungus. In addition to the known mitochondrial role in pathogenicity, our results demonstrated that the global function analysis of this organelle is similar in pathogenic and non-pathogenic fungi, suggesting that its relevance in the lifestyle of these organisms should be based on a small number of specific proteins and/or with respect to differential gene regulation. In this regard, particular interest should be directed towards mitochondrial proteins with unknown function and the novel protein that might be specific to this species. Further functional characterization of these proteins could enhance our understanding of the role of mitochondria in phytopathogenicity.
Related collections
Most cited references72
- Record: found
- Abstract: found
- Article: not found
The Pfam protein families database.
- Record: found
- Abstract: found
- Article: not found