- Record: found
- Abstract: found
- Article: found
An overview of the Phalaenopsis orchid genome through BAC end sequence analysis
Read this article at
Abstract
Background
Phalaenopsis orchids are popular floral crops, and development of new cultivars is economically important to floricultural industries worldwide. Analysis of orchid genes could facilitate orchid improvement. Bacterial artificial chromosome (BAC) end sequences (BESs) can provide the first glimpses into the sequence composition of a novel genome and can yield molecular markers for use in genetic mapping and breeding.
Results
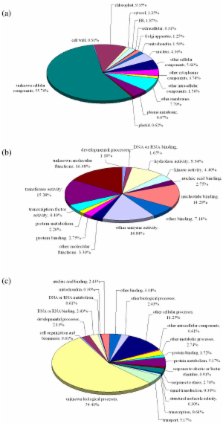
We used two BAC libraries (constructed using the BamHI and HindIII restriction enzymes) of Phalaenopsis equestris to generate pair-end sequences from 2,920 BAC clones (71.4% and 28.6% from the BamHI and HindIII libraries, respectively), at a success rate of 95.7%. A total of 5,535 BESs were generated, representing 4.5 Mb, or about 0.3% of the Phalaenopsis genome. The trimmed sequences ranged from 123 to 1,397 base pairs (bp) in size, with an average edited read length of 821 bp. When these BESs were subjected to sequence homology searches, it was found that 641 (11.6%) were predicted to represent protein-encoding regions, whereas 1,272 (23.0%) contained repetitive DNA. Most of the repetitive DNA sequences were gypsy- and copia-like retrotransposons (41.9% and 12.8%, respectively), whereas only 10.8% were DNA transposons. Further, 950 potential simple sequence repeats (SSRs) were discovered. Dinucleotides were the most abundant repeat motifs; AT/TA dimer repeats were the most frequent SSRs, representing 253 (26.6%) of all identified SSRs. Microsynteny analysis revealed that more BESs mapped to the whole-genome sequences of poplar than to those of grape or Arabidopsis, and even fewer mapped to the rice genome. This work will facilitate analysis of the Phalaenopsis genome, and will help clarify similarities and differences in genome composition between orchids and other plant species.
Conclusion
Using BES analysis, we obtained an overview of the Phalaenopsis genome in terms of gene abundance, the presence of repetitive DNA and SSR markers, and the extent of microsynteny with other plant species. This work provides a basis for future physical mapping of the Phalaenopsis genome and advances our knowledge thereof.
Related collections
Most cited references40
- Record: found
- Abstract: found
- Article: not found
The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla.
- Record: found
- Abstract: found
- Article: not found
Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.).
- Record: found
- Abstract: not found
- Article: not found