- Record: found
- Abstract: found
- Article: not found
alpha2delta expression sets presynaptic calcium channel abundance and release probability
research-article
13 May 2012

There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Synaptic neurotransmitter release is driven by Ca2+ influx through active zone voltage-gated
calcium channels (VGCCs)
1,2
. Control of active zone VGCC abundance and function remains poorly understood. We
show that a trafficking step likely sets synaptic VGCC levels as overexpression of
the pore-forming α1A fails to change synaptic VGCC abundance or function. α2δs are
a family of GPI-anchored VGCC-associated subunits
3
, which in addition to being the target of the potent neuropathic analgesics gabapentin
and pregabalin (α2δ-1, α2δ-2)
4,5
, were also identified in a forward genetic screen for pain genes (α2δ-3)
6
. We show that these proteins confer powerful modulation of presynaptic function through
two distinct molecular mechanisms. α2δ subunits set synaptic VGCC abundance, as predicted
from their chaperone-like function when expressed in non-neuronal cells
3,7
. Secondarily, α2δs configure synaptic VGCCs to drive exocytosis through an extracellular
metal ion-dependent adhesion site (MIDAS), a conserved set of amino acids within α2δ’s
predicted von Willebrand A (VWA) domain. Expression of α2δ with an intact MIDAS motif
leads to an 80% increase in release probability, while simultaneously protecting exocytosis
from blockade by an intracellular Ca2+ chelator. α2δs harboring MIDAS site mutations
still drive synaptic accumulation of VGCCs however, they no longer change release
probability or sensitivity to intracellular Ca2+ chelators. Our data reveal dual functionality
of these clinically important VGCC subunits, allowing synapses to make more efficient
use of Ca2+ entry to drive neurotransmitter release.
VGCCs are composed of pore-forming α1 and auxiliary β and α2δ subunits
8,9
. In central synapses neurotransmitter release is generally driven by P/Q-type (α1A)
and/or N-type (α1B)10 VGCCs. Based on the failure of α1A overexpression to increase
synaptic strength, it had been suggested that VGCCs functionally coupled to presynaptic
release machinery is limited by a fixed number of available “slots” where channels
can insert into the synaptic membrane
11
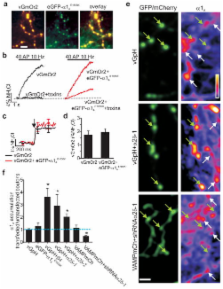
. We examined the existence of such a bottleneck by expressing eGFP-α1A12 together
with a reporter of presynaptic exocytosis (vGlut1 with a luminal tag mOrange2, vGmOr2)
and carried out retrospective immunocytochemistry to probe the abundance of α1A in
transfected compared to control neurons. eGFP-α1A correctly trafficked to nerve terminals
as it co-localized well with the vesicle-targeted reporter (Fig. 1a). In order to
ensure eGFP-α1A functionally integrated with endogenous channels to drive neurotransmitter
release we introduced a point mutation (E1656K), rendering this channel insensitive
to the antagonist ω-agatoxin IVA
13
. Under control conditions a combination of ω-agatoxin IVA and the α1B inhibitor ω-conotoxin
GVIA completely blocked vGmOr2 responses to action potential (AP) firing, however
in the presence of eGFP-α1A
E1656K a significant fraction of the response remains (Fig 1b). Measurements of single
AP responses showed that expression of this exogenous α1A did not alter exocytosis
efficiency compared to controls (Fig. 1c–d), consistent with the “slot” hypothesis
11
. However, retrospective immunocytochemistry using an anti-α1A antibody whose specificity
was verified using shRNA-mediated α1A knockdown (Fig. S1) showed that transfected
and control nerve terminals had similar immunoreactivity (Fig. 1e–f) while at the
cell soma it had doubled (Fig S2). These results demonstrate that synaptic VGCC abundance
is likely limited by trafficking from the cell soma and failure to increase synaptic
performance does not result from a fixed number of active zone insertion sites. α2δ
and β auxiliary VGCC subunits are both strong candidates for modulating such trafficking
as they control functional expression of α1 subunits when co-expressed in non-neuronal
cells
14,15
. We coexpressed individual auxiliary subunits with the reporter vGlut1-pHluorin (vGpH)
in neurons and carried out measurements of exocytosis and immunocytochemistry as described
above. These experiments demonstrated that expression of either α2δ-1 or β4 subunits
led to a significant increase (~3-fold, p>0.05) in synaptic abundance of α1A (Fig.
1e–f). Similar results were obtained with overexpression of α2δ-2 (Fig. 1f). Furthermore,
introduction of shRNA targeting α2δ-1 caused depletion of α1A at nerve terminals (Fig.
1e–f, Figure S3), while leaving the somatic concentration unaltered (data not shown).
These results demonstrate that synaptic α1A levels are titrated by expression of auxiliary
VGCC subunits.
To examine whether changes in VGCC accumulation alters synaptic release properties,
we measured single AP-stimulated exocytosis in neurons with altered VGCC levels. Overexpression
and depletion of α2δ–1 led to much larger and much smaller single AP responses respectively
compared to control (Fig. 2a). Similar increases in exocytosis were observed following
expression of all three isoforms of α2δ tested (Fig. 2b). In contrast, expression
of β4 did not change exocytosis, despite the synaptic accumulation of α1A (Fig. 2b).
Quantitative estimates of α2δ-1 synaptic expression levels suggest a stoichiometric
relationship between α2δ and α1A (Fig. S4). These results demonstrate that increasing
VGCC abundance does not necessarily lead to increased function, and identify α2δ expression
as a key rate-limiting parameter in determining presynaptic function.
Measurements of presynaptic strength can be parsed into two biophysical parameters:
the number of vesicles available for rapid release upon stimulation, known as the
readily-releasable pool (RRP) and the probability that a vesicle in the RRP will undergo
fusion with a single AP stimulus (Pv)
16
. We recently developed a rapid depletion protocol to measure RRP sizes using optical
methods
17
. High frequency stimulation leads to rapid exhaustion of exocytosis in the first
8–15 APs. The fraction of the total pool corresponding to this rapid depletion phase
is taken as the RRP
17
(Fig. 2c–d). The RRP size in neurons overexpressing α2δ was no different than controls
(Fig. 2e). Thus α2δ overexpression changes Pv (Fig. 2f).
α2δ-driven increases in Pv might arise from increasing total Ca2+ influx (from changes
in VGCC gating and/or surface abundance) and/or changing VGCC proximity to release
sites
18
. To examine this question, we measured intracellular [Ca2+] at synaptic boutons in
response to single APs using the fast fluorescent indicator of Ca2+, Fluo5F-AM, visualized
by expression of VAMP-mCherry (VAMPmCh; Fig. 3a left panel) with or without α2δ. Single
APs resulted in robust Ca2+ signals (Fig. 3a right panel) that peaked within 1 ms.
but were reduced by ~40% in synapses overexpressing α2δ isoforms compared to controls
(Fig. 3b–c). We verified that the peak signal was not dominated by Ca2+ clearance
mechanisms (e.g. endogenous buffers or extrusion) in experiments where the signal
decay was set by high concentrations of intracellular EGTA. This treatment led to
a ~50% decrease in peak signal and a decay time of ~10 ms in controls as well as α2δ
overexpressing synapses. Measurements of Ca2+ signals using a genetically encoded
Ca2+ indicator GCaMP3
19
, co-expressed with or without α2δ-1 gave very similar results (Fig. S5). This reduction
in Ca2+ was surprising given that α2δ overexpression increases the total number of
synaptic VGCCs (surface and intracellular) suggesting that α2δ might additionally
control Ca2+ influx. Measurements of somatic AP waveforms revealed that α2δ expression
led to a ~30% decrease in AP duration (Fig. S6), providing a possible explanation
for the drop in synaptic Ca2+ entry. Given that exocytosis at nerve terminals is steeply
dependent on Ca2+ influx
20
, the proximity of sites of Ca2+ influx to sites of exocytosis can, in principle,
have a powerful influence over neurotransmitter release
2
. The increase in Pv with a commensurate decrease in Ca2+ influx strongly suggests
that overexpression of α2δ subunits results in a tighter spatial relationship between
sites of Ca2+ entry and exocytosis. We tested this hypothesis by measuring the sensitivity
of exocytosis to the presence of a Ca2+ chelator (EGTA-AM). The chelator’s efficiency
in reducing exocytosis depends on its ability to buffer Ca2+ before it binds the calcium-sensor
for exocytosis following VGCC opening, a process determined by chelator concentration
and Ca2+ binding kinetics
21
. We chose incubation conditions for EGTA-AM that led to a ~50% reduction in single
AP exocytosis responses, compared to the pre-EGTA condition in control neurons (Fig.
3d). In neurons transfected with α2δ however, EGTA application led to much smaller
decreases in exocytosis (Fig. 3e) indicating that in conditions of α2δ overexpression
Ca2+ must bind the calcium sensor more rapidly than in control conditions. Therefore
the Ca2+ sensor controlling exocytosis is experiencing higher levels of Ca2+ influx
even though overall synaptic Ca2+ transients are reduced. Single AP-driven Ca2+ influx
remained equally sensitive to ω-conotoxin GVIA following α2δ overexpression indicating
this condition did not lead to a significant shift in VGCC type at nerve terminals
(Fig. S7).
The finding that α2δ subunits form GPI-anchored proteins
3
implies that their ability to change VGCC-exocytosis coupling is likely conveyed through
an extracellular interaction. One possible candidate for exerting such influence lies
in the highly-conserved VWA domain within α2δ
22
. A characteristic feature of this domain is its ability to interact with adhesion
proteins via the MIDAS motif by sharing coordination of a divalent cation
23–25
. To examine the role of α2δ’s MIDAS motif we mutated three of five conserved key
metal coordinating residues within the MIDAS motif to alanine
22
and expressed the mutant protein (α2δ-1 MIDASAAA) in neurons together with functional
reporters. α2δ-1 MIDASAAA was similar to wild type α2δ-1 in its ability to drive α1A
accumulation at synapses (Fig. 4a). However, measurements of exocytosis from α2δ-1
MIDAS-mutants showed no enhancement of Pv (Fig. 4b), normal Ca2+ influx (Fig. 4c)
and normal sensitivity to EGTA block of exocytosis (Fig. 4d). Furthermore α2δ-2 MIDASAAA,
unlike intact α2δ-2, was unable to rescue the decrease in exocytosis resulting from
shRNA-mediated α2δ-1 depletion (Fig. 4e,f). These data are consistent with the ability
of this mutation to block enhancement of Ca2+ currents when expressed in heterologous
systems
22
(Fig. S8), but show that they do not prevent endogenous α2δ from functioning. Taken
together, these results demonstrate that α2δ exerts its powerful control of synaptic
VGCC function through at least two separate molecular mechanisms: a forward trafficking-step
from cell body to presynaptic terminal that is independent of MIDAS motif integrity;
and a local MIDAS-dependent interaction critical for proper VGCC function and coupling
to exocytosis.
α2δ-1 and α2δ-2 are the targets of the analgesic gabapentin whose binding site lies
in close proximity to the MIDAS site
26
. We found no significant impact of gabapentin application (30 min and >72 h) on Pv
in either control or cells overexpressing α2δ-1 or -2 (results not shown) similar
to previous findings in hippocampal neurons
27
. We also examined gabapentin’s impact on VGCC trafficking to nerve terminals by incubating
neurons with gabapentin from the time of transfection with eGFP-α1A. Analysis of the
presynaptic abundance of this probe after 7 days showed that even though gabapentin
appears to impact α2δ-2 trafficking in non-neuronal cells
28
it appears unable to impact VGCC trafficking or function in cultured hippocampal neurons
(Fig. S9).
Our results reveal that α2δ subunits are potent modulators of synaptic transmission.
They function through at least two distinct molecular mechanisms: a trafficking step
from the cell soma and a local step at the presynaptic terminal allowing synapses
to exhibit increased exocytosis with decreased Ca2+ influx. We speculate that increased
presynaptic abundance of VGCCs results in increased abundance of active zone-VGCCs,
and hence a higher density in the vicinity of release sites. This active zone accumulation
depends on α2δ’s VWA domain, presumably through interactions with extracellular active
zone-specific proteins. The identity of the interaction partner(s) at present remains
unknown however it is tempting to speculate that α2δs might recognize cues established
in the correct juxtaposition of pre and postsynaptic membranes, consistent with synaptic
defects observed in Drosophila α2δ-3 mutants
29
. Additionally this model requires that α2δ interact with a partner resulting in AP
shortening to limit total calcium entry. As the MIDAS motif is well conserved throughout
the α2δ family, identification of α2δ interaction partners in specific neuronal circuits
could provide novel targets in the development of future therapeutics, given the potency
that these subunits show in controlling synapse function.
Methods
Hippocampal CA3–CA1 regions were dissected from 1- to 3-day-old Sprague Dawley rats,
dissociated, plated and transfected as previously described
30
. Live-cell images were acquired with an Andor iXon+ (model #DU-897E-BV) camera. A
solid-state diode pumped 488 nm (vGpH and MgG imaging) or 532 nm (vGmOr2 imaging)
laser was shuttered using acousto-optic modulation. For vGpH and GCaMP3 imaging data
were acquired at 100 Hz by integrating for 9.74 ms in frame transfer mode and restricting
imaging to a sub-area of the CCD chip. Fluo5F imaging data was acquired at 1 kHz (0.974
ms integration time). To estimate 1 AP ΔFs of vGpH, we took the difference between
the average 20 frames before and after the stimulus. The rise in vGpH fluorescence
in response to a single AP always took two frames when acquiring at 100 Hz time resolution.
For display purposes the images in Fig. 1E were given a 2 pixel gaussian average filter.
Pv and single AP calcium signals were measured with 4 mM extracellular CaCl2. All
stated values are mean±SEM, statistical significance for groups of 3 or more were
determined by one-way ANOVA with Tukey’s HSD for Post-Hoc analysis. Otherwise Student’s
t-test was used for determining statistics.
Supplementary Material
1
2
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: not found
Multiple roles of calcium ions in the regulation of neurotransmitter release.
Erwin Neher, Takeshi Sakaba (2008)
- Record: found
- Abstract: found
- Article: not found
Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere.
Charles A Whittaker, Richard O. Hynes (2002)
- Record: found
- Abstract: found
- Article: not found
Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin.
J Offord, Laura Corradini, Ian A. Kinloch … (2006)