- Record: found
- Abstract: found
- Article: found
Ecological Drivers of the Soil Microbial Diversity and Composition in Primary Old-Growth Forest and Secondary Woodland in a Subtropical Evergreen Broad-Leaved Forest Biome in the Ailao Mountains, China
Read this article at
Abstract
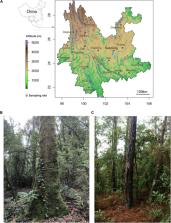
Replacement of primary old-growth forests by secondary woodlands in threatened subtropical biomes drives important changes at the level of the overstory, understory and forest floor, but the impact on belowground microbial biodiversity is yet poorly documented. In the present study, we surveyed by metabarcoding sequencing, the diversity and composition of soil bacteria and fungi in the old-growth forest, dominated by stone oaks ( Lithocarpus spp.) and in the secondary Yunnan pine woodland of an iconic site for biodiversity research, the Ailaoshan National Nature Reserve (Ailao Mountains, Yunnan province, China). We assessed the effect of forest replacement and other environmental factors, including soil horizons, soil physicochemical characteristics and seasonality (monsoon vs. dry seasons). We showed that tree composition and variation in soil properties were major drivers for both bacterial and fungal communities, with a significant influence from seasonality. Ectomycorrhizal Operational Taxonomic Units (OTUs) dominated the functional fungal guilds. Species richness and diversity of the bacterial and fungal communities were higher in the pine woodland compared to the primary Lithocarpus forest, although prominent OTUs were different. The slightly lower complexity of the microbiome in the primary forest stands likely resulted from environmental filtering under relatively stable conditions over centuries, when compared to the secondary pine woodlands. In the old-growth forest, we found a higher number of species, but that communities were homogeneously distributed, whereas in the pine woodlands, there is a slightly lower number of species present but the communities are heterogeneously distributed. The present surveys of the bacterial and fungal diversity will serve as references in future studies aiming to assess the impact of the climate change on soil microbial diversity in both old-growth forests and secondary woodlands in Ailaoshan.
Related collections
Most cited references63

- Record: found
- Abstract: found
- Article: found
edgeR: a Bioconductor package for differential expression analysis of digital gene expression data
- Record: found
- Abstract: found
- Article: not found
DADA2: High resolution sample inference from Illumina amplicon data

- Record: found
- Abstract: found
- Article: found