- Record: found
- Abstract: found
- Article: found
tRNA Signatures Reveal a Polyphyletic Origin of SAR11 Strains among Alphaproteobacteria
Read this article at
Abstract
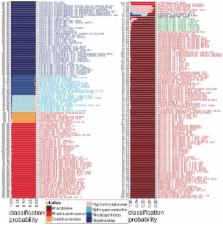
Molecular phylogenetics and phylogenomics are subject to noise from horizontal gene transfer (HGT) and bias from convergence in macromolecular compositions. Extensive variation in size, structure and base composition of alphaproteobacterial genomes has complicated their phylogenomics, sparking controversy over the origins and closest relatives of the SAR11 strains. SAR11 are highly abundant, cosmopolitan aquatic Alphaproteobacteria with streamlined, A+T-biased genomes. A dominant view holds that SAR11 are monophyletic and related to both Rickettsiales and the ancestor of mitochondria. Other studies dispute this, finding evidence of a polyphyletic origin of SAR11 with most strains distantly related to Rickettsiales. Although careful evolutionary modeling can reduce bias and noise in phylogenomic inference, entirely different approaches may be useful to extract robust phylogenetic signals from genomes. Here we develop simple phyloclassifiers from bioinformatically derived tRNA Class-Informative Features (CIFs), features predicted to target tRNAs for specific interactions within the tRNA interaction network. Our tRNA CIF-based model robustly and accurately classifies alphaproteobacterial genomes into one of seven undisputed monophyletic orders or families, despite great variability in tRNA gene complement sizes and base compositions. Our model robustly rejects monophyly of SAR11, classifying all but one strain as Rhizobiales with strong statistical support. Yet remarkably, conventional phylogenetic analysis of tRNAs classifies all SAR11 strains identically as Rickettsiales. We attribute this discrepancy to convergence of SAR11 and Rickettsiales tRNA base compositions. Thus, tRNA CIFs appear more robust to compositional convergence than tRNA sequences generally. Our results suggest that tRNA-CIF-based phyloclassification is robust to HGT of components of the tRNA interaction network, such as aminoacyl-tRNA synthetases. We explain why tRNAs are especially advantageous for prediction of traits governing macromolecular interactions from genomic data, and why such traits may be advantageous in the search for robust signals to address difficult problems in classification and phylogeny.
Author Summary
If gene products work well in the networks of foreign cells, their genes may transfer horizontally between unrelated genomes. What factors dictate the ability to integrate into foreign networks? Different RNAs and proteins must interact specifically in order to function well as a system. For example, tRNA functions are determined by the interactions they have with other macromolecules. We have developed ways to predict, from genomic data alone, how tRNAs distinguish themselves to their specific interaction partners. Here, as proof of concept, we built a robust computational model from these bioinformatic predictions in seven lineages of Alphaproteobacteria. We validated our model by classifying hundreds of diverse alphaproteobacterial taxa and tested it on eight strains of SAR11, a phylogenetically controversial group that is highly abundant in the world's oceans. We found that different strains of SAR11 are more distantly related, both to each other and to mitochondria, than widely believed. We explain conflicting results about SAR11 as an artifact of bias created by the variability in base contents of alphaproteobacterial genomes. While this bias affects tRNAs too, our classifier appears unexpectedly robust to it. More broadly, our results suggest that traits governing macromolecular interactions may be more faithfully vertically inherited than the macromolecules themselves.
Related collections
Most cited references67
- Record: found
- Abstract: found
- Article: not found
The Bioperl toolkit: Perl modules for the life sciences.
- Record: found
- Abstract: found
- Article: not found
Fast UniFrac: Facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data
- Record: found
- Abstract: found
- Article: not found