
INTRODUCTION
Recent development of time-resolved X-ray scattering techniques have allowed for detection of the structure of several transient species employing a laser-pump and an X-ray scattering-probe [1–6]. Tracking nuclear motion in a solvent system enables the structural description of transients during the progression of a chemical reaction. The requirements for the systems that are to be investigated are strict. The lifetime of the transient species must be known, a large concentration of the species must be generated and the species must scatter X-rays significantly different from the species present at equilibrium. In this article, potential candidates for the study of transients in a 2 + 2 cycloaddition reaction with time-resolved X-ray scattering studies in solution and the gas phase are presented.
The norbornadiene-quadricyclane system has been investigated as a potential system for solar energy storage [7–12]. The system has been a topic of intense interest [13–19], and a mechanism for the conversion from norbornadiene to quadricyclane has been proposed [7, 15, 20–29]. The mechanism is shown in Figure 1 and involves two transient species with different structures. If the mechanism is correct, the system will display a disappearance of N, the grow-in and disappearance of 3DN and 3DQ, and the finally grow-in of Q if investigated using a pump-probe technique capable of determining structural change.

Simulations employing structure minimisation and calculation of scattering patterns show that the structure of the different species can indeed be resolved by X-ray scattering if heavy atoms are substituted onto the system (assuming an overall quantum yield of 10% for the process). Furthermore, we speculated that if the heavy atoms were introduced in a conjugated system they would bring the systems absorption energy out of the ultraviolet (UV) region, minimising the chance of photoinduced carbon-bromine homolytic bond dissociation.
Two new norbornadienes were prepared with two and four bromine substituents, respectively. By absorption and NMR spectroscopy, both systems were shown to reversibly form the corresponding quadricyclanes.
RESULTS AND DISCUSSION
The synthetic route to 5,6-bis(4-bromophenyl)-7,7-dimethyl-norbornadiene-2,3-dicarboxylic acid (8) and the corresponding bis(4-bromophenyl) ester (9) is shown in Scheme 1. The starting material 1 [30] was converted to the key intermediate: 2-hydroxy-4,4-dimethylcyclopent-2-enone (3), by an acyloin condensation [31–32]. The sodium/potassium alloy used in this step is highly flammable and limits the reaction scale to 10 g. Two sequential reactions with 4-bromphenyllithium followed by dehydration with 4-toluenesulfonic acid give 2,3-bis(4-bromphenyl)-5,5-dimethyl cyclopentadiene (7) in an overall yield of 26%. Acetylenedicarboxylic and 7 underwent a Diels-Alder condensation in excellent yields to form the norbornadiene acid 8, which was made into the norbornadiene ester 9 via the acid chloride and 4-bromophenol. The multi-step synthesis gave 8 and 9 in acceptable overall yields of 17% and 13%, respectively.

The molecular structure of the sodium salt of 8 as crystallised from acetone was confirmed by single-crystal X-ray diffraction (CCDC 817053) and is shown in Figure 2. The structure clearly reveals the locked stilbene system that the bromine atoms are placed in. The aromatic rings may rotate, but the positions of the bromine atoms are fixed. In 9, the bromine atoms in the ester groups have a higher degree of freedom and more conformers are possible. Even so, the structural changes when norbornadiene becomes quadricyclane will result in systematic changes in the interatomic distances, in particular for the bromine atoms in 8 and 9. The bromine atoms contribute very significantly to the X-ray scattering signature of the systems by introducing a strong modulation in the electron density distribution. Hence, these changes can be monitored using time-resolved X-ray scattering techniques. The bromine atoms account for more than 25% of the total electrons of the system.

If the norbornadienes 8 and 9 are to be studied using ultrafast X-ray scattering techniques, it must first be shown that the norbornadienes 8 and 9 can undergo photolytic isomerisation to the corresponding quadricyclanes 8q and 9q As triplet states are involved in the transformation [7–12], the bromine atoms might make the process slow or prevent it from happening. Two photolysis experiments are shown in Figure 3. It clearly shows a reaction happening. A NMR investigation of the photolysis product confirms that the quadricyclane is the only product formed. The photolysis was carried out for 8 and 9 using 290 nm and 350 nm light in several solvents (Benzene, CHCl3, CH3CN, CH3OH). Although, the absorption is strongest at 290 nm, the produced quadricyclanes also absorb light at this wavelength. Hence, 350 nm light was used in the experiments shown here.

The effect of triplet sensitisers was tested using identical conditions as those used to generate the data in Figure 3. No effect of the sensitisers was found. The data are in all cases identical to those shown in Figure 3 when the sensitiser absorbance has been subtracted. The bromine atoms seem to act as internal triplet sensitisers and as such allow for an efficient conversion from norbornadiene to quadricyclane.
The photolysis was also carried out in deuterated solvents in order to perform NMR experiments. Using NMR, it was determined that the photostationary state at 350 nm gave a ratio of [Q]/[N] = 92:8 in chloroform and 90:10 in benzene.
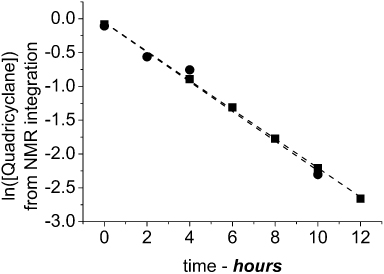
In most cases, the reverse process, from quadricyclane to norbornadiene, has to be catalysed [26, 33]. Furthermore, the quadricyclane formation is not fully reversible in the known systems [34]. In both 8/8q and 9/9q, the photolysis is fully thermally reversible. Figure 4 shows a time-lapse NMR experiment in deutero-chloroform. It shows the thermal regeneration of norbornadiene over 12 hours at room temperature. The sample is kept in the dark.

Figure 5 shows the kinetic traces generated from the NMR experiments in deutero-chloroform and deutero-benzene. The t½ = 3 hours and the half-life is the same for all investigated solvents and the same for both systems 8/8q and 9/9q. After 24 hours, no traces of quadricyclane or other impurities remain. Thus, the system can be cycled repeatedly between norbornadiene and quadricyclane without loss of material.
CONCLUSIONS
Two new norbornadienes with bromine substituents have been synthesised and characterised. They are both capable of photolytic valence isomerisation to quadricyclane and can thermally be converted back to norbornadiene. The full recovery of the norbornadiene, combined with the unsensitised photoactivated isomerisation, make these systems good candidates for time-resolved X-ray studies using pump-probe techniques. The structural changes when going from norbornadiene over the relevant intermediates to quadricyclane are accompanied by the significant changes in electron density distribution due to the bromine atoms and can therefore be probed directly by X-ray scattering.
EXPERIMENTAL SECTION
Unless otherwise stated, all starting materials were obtained from commercial suppliers and used as received. Solvents were of HPLC grade and were used as received. Ground-state absorption spectroscopy was routinely recorded with a Cary 100 Bio spectrophotometer as solutions in the stated solvents. 1H NMR and 13C NMR spectra were recorded on a 400 MHz (Varian) instrument (400 MHz for 1H NMR and 100 MHz for 13C NMR) or on a 500 MHz (Varian) instrument (500 MHz for 1H NMR and 125 MHz for 13C NMR). Proton chemical shifts are reported in ppm downfield from tetramethylsilane (TMS) and carbon chemical shifts in ppm downfield of TMS, using the resonance of the solvent as internal standard. Melting points were measured on a Gallenkamp apparatus and are uncorrected. High resolution mass spectrometry (HRMS) were recorded on a Micromass Q-TOF apparatus using electrospray ionisation (ESI) technique. Matrix assisted laser desorption ionisation time of flight (MALDI-TOF) mass spectra were recorded on a VG TofSpec spectrometer. Electrospray ionisation (ESI) was recorded on a ThermoQuest Finnigan LCQ DECA instrument. Dry column vacuum chromatography was performed on Merck Kiselgel 60 (0.015–0.040 mm) and gravity feed column chromatography was performed on Merck Kiselgel 60 (0.040–0.063 mm). Thin layer chromatography (TLC) was carried out using aluminium sheets pre-coated with silica gel 60F (Merck 5554).
4,4-Dimethyl-1,2-cyclopentadione/2-hydroxy-4,4-dimethylcyclopent-2-enone (3). A flame-dried 2-L tree-necked round-bottom flask equipped with a reflux condenser, addition funnel and a mechanical stirrer was maintained under an oxygen-free, nitrogen atmosphere. The flask is charged with freshly cut sodium (60 g) and freshly cut potassium (12 g), and the flask is heated with a heat gun, forming the low-melting alloy. Dry benzene (1200 mL) is added, and the stirrer is operated at high speed until the alloy is dispersed and then at a slower speed for the remaining reaction time. The dispersion was kept at 25°C and diester 1 (21.4 g, 0.11 mol), and Me3SiCl (87.6 g, 0.8 mol) was dissolved in dry benzene (100 mL) and added carefully through the addition funnel. After 20 hours with rapid stirring, the mixture was filtered through a plug of celite under an argon atmosphere and concentrated in vacuo to give a colourless oil of compound 2. TLC (50% CH2Cl2/Hexane) Rf = 0.8; Gas chromatography mass spectrometry (GCMS) (m/z (intensities)): 272 (100), 147 (44), 73 (85). The oil was dissolved in dry CH2Cl2 (500 mL) at −78°C under an N2 atmosphere, and Br2 (17.8 g, 1.1 mol) was added dropwise over a 5-minute period, and the reaction was allowed to warm to 25°C. The mixture was concentrated in vacuo. The product was purified by dry column vacuum chromatography (from heptane to EtOAc-heptane with 10% increments). Yield: 10.1 g, 72%; TLC (50% CH2Cl2/Hexane) Rf = 0.8; 1H NMR (CDCl3, 300 MHz): δ6.41 (s, 1H), 6.23 (s, 1H), 2.30 (s, 2H), 1.25 (s, 6H); GCMS (m/z (intensities)): 126 (35), 111 (100), 83 (45), 55 (45), 43 (50).
2-(4-Bromophenyl)-2-hydroxy-4,4-dimethylcyclopentanone (4). A flame-dried 50-mL round-bottom flask equipped with a rubber septum was added p-Dibromobenzene (3.54 g, 15 mmol) dissolved in a mixture of THF (30 mL) and diethyl ether (30 ml). n-Butyllithium (2.5 M in hexane, 6 ml, 15 mmol) was added dropwise using a syringe at −78°C under a nitrogen atmosphere. After stirring under argon at −78°C for an additional half an hour, 2-hydroxy ketone 3 (500 mg, 3.96 mmol) dissolved in dry diethyl ether (15 mL) was added dropwise using a syringe, then the mixture was allowed to reach room temperature over a period of 16 hours. The reaction mixture was quenched in ice water and extracted with ether (2 ×50 mL). The extracts was washed with brine (50 mL), dried with MgSO4 and evaporated to dryness in vacuo. Purification by dry column vacuum chromatography (from heptane to EtOAc-heptane with 5% increments, starting by washing with heptane) yielded: 860 mg, 76%; m.p. = 94–95°C; TLC (50% EtOAc/Hexane) Rf = 0.9; 1H NMR (CD3CN, 400 MHz): δ7.51 (d, 2H, J = 8.8 Hz), 7.30 (d, 2H, J = 8.8 Hz), 3.89 (s, 1H), 2.40 (q, 2H, J = 16.8 Hz), 2.17 (m, 2H), 1.23 (s, 3H), 1.16 (s, 3H); 13C NMR (CDCl3, 100 MHz,): δ217.2, 143.5, 131.4, 127.9, 120.9, 81.2, 53.5, 52.8, 33.1, 29.6, 28.9; GCMS (m/z (intensities)): 200 (97), 198 (100), 185 (52), 183 (55); Anal. Calcd. For C13H15BrO2: C, 55.14; H, 5.35; Found: C, 54.16; H, 5.35.
2-(4-Bromophenyl)-4,4-dimethylcyclopent-2-enone (5). In a 100-mL three-necked round-bottom flask equipped with a magnetic stirrer, a reflux condenser was added a solution of hydroxy-cyclopentanone 4 (3.0 g, 10.6 mmol) dissolved in toluene (120 mL) and TsOH·H2O (2.0 g, 10.8 mmol) was added. The mixture was heated to reflux for 40 minutes. The reaction mixture was allowed to cool to room temperature and then poured into 10% NaOH solution (100 mL) and extracted with diethyl ether (2 × 50 mL). The extracts were washed with brine (100 mL), dried with MgSO4 and evaporated to dryness in vacuo. Purification by dry column vacuum chromatography (from heptane to EtOAc-heptane with 10% increments) yielded: 2.0 g, 71%; m.p. = 65–67°C; TLC (20% EtOAc/Hexane) Rf = 0.5; 1H NMR (CDCl3, 500 MHz): δ7.60 (d, 2H, J = 8.5 Hz), 7.58 (s, 1H), 7.59 (d, 2H, J = 8.5 Hz), 2.47 (s, 2H), 1.30 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 207.08, 168.25, 139.33, 131.76, 130.43, 128.97, 122.74, 51.62, 38.55, 28.43; GCMS (m/z (intensities)): 266 (53), 264 (55), 251 (42), 249 (44), 157 (40), 142 (100); Anal. Calcd. For C13H13BrO: C, 58.89; H, 4.94; Found: C, 58.65; H, 5.02.
1,2-Bis(4-bromophenyl)-4,4-dimethylcyclopent-2-en-1-ol (6). A flame-dried 50-mL round-bottom flask equipped with a rubber septum was added p-dibromobenzene (3.54 g, 15 mmol) dissolved in a mixture of dry THF (30 mL) and dry diethyl ether (30 mL). n-Butyllithium (2.5 M in hexanes, 5 mL, 1.3 mmol,) was added dropwise using a syringe at –78°C under a nitrogen atmosphere. After stirring under argon at –78°C for an additional half hour, cyclopentenone 5 (1.00 g, 3.77 mmol) dissolved in dry diethyl ether (15 mL) was added dropwise using a syringe and then the mixture was allowed to reach room temperature over a period of 16 hours. The reaction mixture was quenched in a ice water (200 mL) and extracted with ether (2 × 100 mL). The extracts were washed with brine, dried with MgSO4 and evaporated to dryness in vacuo. Purification by dry column vacuum chromatography (from heptane to EtOAc-heptane with 50% increments, starting by washing with heptane) yielded: 1.2 g, 75%; m.p. = 103–106°C; TLC (20% EtOAc/Hexane) Rf = 0.7; 1H NMR (CD3CN, 400 MHz): δ1H NMR (400 MHz, CDCl3) δ7.31 (d, 2H, J = 8.8 Hz), 7.21 (m, 4H), 7.13 (d, 2H, J = 8.8 Hz), 6.22, (s, 1H), 2.16 (d, 2H, J = 4.2 Hz), 1.22 (s, 3H), 1.09 (s, 3H); 13C NMR (CDCl3, 101 MHz) δ146.04, 143.41, 141.87, 132.96, 131.56, 131.51, 128.96, 127.37, 121.49, 120.71, 88.35, 77.58, 77.26, 76.94, 60.64, 42.90, 30.81, 29.11; GCMS (m/z(intensities)): 422 (45), 225 (54), 223 (58), 185 (98), 183 (100), 128 (64); Anal. Calcd. For C19H18Br2O: C, 50.06; H, 4.30; Found: C, 50.09; H, 4.38.
2,3-bis(p-bromphenyl)-5,5-dimethyl cyclopentadiene (7). In a 100-mL three-necked round-bottom flask equipped with a magnetic stirrer, a reflux condenser was added a solution of hydroxyl-cyclopentenone 6 (1.2 g, 2.4 mmol) dissolved in toluene (50 mL) and TsOH·H2O (480 mg, 2.5 mmol) was added. The mixture was heated to reflux for 40 minutes. The mixture were poured into 10% NaOH solution and extracted with diethyl ether (3 × 50 mL). The extracts were washed with brine (100 mL), dried with MgSO4 and evaporated to dryness in vacuo. Purification by dry column vacuum chromatography (heptane) yielded: 740 mg, 64%; m.p. = 158–159°C; TLC (20% toluene/Hexane) Rf = 0.7; 1H NMR (CDCl3, 400 MHz): δ7.36 (d, 4H, J = 8.6 Hz), 6.98 (d, 4H, J = 8.6 Hz), 6.34 (s, 2H), 1.31 (s, 6H); 13C NMR (CDCl3, 101 MHz,): δ145.4, 140.4, 134.6, 130.6, 129.2, 120.4, 50.5, 21.9; GCMS (m/z(intensities)): 404 (100), 229 (58), 228 (52); Anal. Calcd. For C19H16Br2: C, 56.47; H, 3.99; Found: C, 56.36; H, 4.03.
5,6-bis(4-bromophenyl)-7,7-dimethylnorbornadiene-2,3-dicarboxylic acid (8). Acetylenedicarboxylic acid (23 mg, 0.2 mmol) was added to a stirring solution of cyclopentadiene 7 (50 mg, 0.124 mmol) dissolved in toluene (20 mL). After stirring for 8 hours at reflux under an N2 atmosphere, the resulting colourless solution was concentrated in vacuo. The residue was subjected to dry column vacuum chromatography (from heptane to EtOAc-heptane with 10% increments) yielded light yellow crystals. Yield: 62 mg, 96%; m.p. = ∼160°C (decomp.); TLC (EtOAc) Rf = 0.1; 1H NMR (500 MHz, CD3OD) δ 7.22 (d, J = 8.3, 2H), 7.03 (d, J = 8.3, 2H), 3.76 (s, 1H), 1.17 (d, J = 12.6, 2H), 1.04 (s, 2H); 13C NMR (CD3OD, 101 MHz) δ 146.34, 135.93, 131.57, 129.10, 121.08, 80.32, 69.68, 21.73, 21.61, 6.42; Anal. Calcd. For C23H18Br2O4 + ½H2O: C, 52.40; H, 3.63; Found: C, 52.48; H, 3.59.
Bis(4-bromphenyl)5,6-bis(4-bromophenyl)-7,7-dimethylnorbornadiene-2,3-dicarboxylate (9). A flame-dried 500 mL tree-necked round-bottom flask equipped with a rubber septum and a reflux condenser was added bis(carboxylic acid) 8 (200 mg, 0.386 mmol) dissolved in dry CH2Cl2 (40 mL). Oxalyl chloride (1.0 ml, 1.1 mmol) was added dropwise using a syringe followed by addition of DMF (cat. Amount 2 drops) at 0°C under a nitrogen atmosphere. The reaction mixture was allowed to warm to room temperature over a period of 1 hour. The reaction mixture was transferred to a 50 mL round-bottom flask and evaporated to dryness in vacuo affording the desired crude acid chloride as a light yellow solid. The acid chloride was used immediately in the next step. To the crude acid chloride was added dry CH2Cl2 (20 mL), Et3N (2 mL) and 4-bromophenol (260 mg, 1.5 mmol), followed by addition of 4-dimethylamino-pyridine (DMAP, cat. Amount 2–4 mg). The mixture was stirred at room temperature for 1 hour and concentrated in vacuo. Purification by dry vacuum chromatography (from heptane to EtOAc with 1% increments) yielded a yellow powder. Yield: 240 mg, 74%. m.p. = 296–298°C; TLC (20% EtOAc/Hexane) Rf = 0.8; 1H NMR (CDCl3, 400 MHz): δ7.40 (d, 4H, J = 8.6 Hz), 7.33 (d, 4H, J = 8.6 Hz), 7.12 (d, 4H, J = 8.5 Hz), 6.90 (d, 4H, J = 8.5 Hz), 3.89 (s, 2H), 1.37 (s, 3 H), 1.35 (s, 3H); 13C NMR (CDCl3, 101 MHz,): δ162.8, 150.8, 149.0, 145.7, 134.9, 132.5, 131.8, 128.8, 123.1, 121.7, 119.3, 81.9, 69.2, 29.6, 22.6; Anal. Calcd. For C35H24Br4O4: C, 50.76; H, 2.92; Found: C, 50.52; H, 3.03; MS (MALDI(TOF)) m/z: calculated for C35H 24Br4O4 + H3O+: 847.2, found: 847.7.