
Monkeypox virus is a rare viral zoonotic orthopoxvirus that causes a disease with symptoms similar to, but less severe than, smallpox. It can be transmitted through body contact, internal mucosal surfaces, or contaminated objects [2, 3]. With the eradication of smallpox in 1980 and the subsequent cessation of vaccination against smallpox, monkeypox became one of the most severe poxviruses. After an incubation period of 5–21 days, monkeypox infection leads to fever, swollen lymph nodes, and an extensive characteristic rash [2, 4]. The documented mortality rate is between 0% and 11%, and has been reported to be higher among young children [5]. Beyond preventing monkeypox through avoiding primary animal-to-human transmission, vaccination may be effective against monkeypox infection [6]. However, some populations in which routine smallpox vaccination has been terminated are more susceptible to monkeypox, because such vaccination provides >80% effective cross-protection against monkeypox [7]. Various compounds against the monkeypox virus are also under development [8].
Two distinct clades have been identified: the West African clade and the Congo Basin clade, also known as the Central African clade [3]. Recently, the genome sequence of the monkeypox virus variant associated with the current outbreak affecting multiple countries has been reported. Rapid phylogenetic analysis has indicated that the 2022 variant belongs to the West African clade and is most closely associated with the variant from Nigeria in 2018. With the increase in monkeypox cases worldwide, better understanding of the new variant is important for accelerating the development of anti-monkeypox vaccines and drugs.
First, we collected the genetic sequences of the well-characterized monkeypox virus variants—the 1996 Congo virus strain (Zaire-96-I-16, ID: NC_003310.1), 2018 West African strain (MPXV-UK_P3, ID: MT903345.1), and 2022 West African strain MPXV_USA_2022_MA001, ID: ON563414.3)—from the NCBI databank to generate whole proteome datasets. We followed the NCBI databank’s open reading frame to extract 191, 190, and 190 proteins from the 1996, 2018, and 2022 strains, respectively. The 2018 West African strain showed a very similar proteome to that of the 2022 strain, with correspondence among all 190 proteins. However, seven proteins in the 1996 strain do not exist in the 2018 and 2022 strains ( Figure 1A ). For example, a gene named BR-209 in the 1996 Congo virus strain encodes a full-length 326 amino acid (A.A.) protein, which is composed of an N-terminal fragment of 210 A.A. and a C-terminal fragment of 126 A.A. However, the West African strains contain a one-base insertion near the N terminus and a four-base deletion, thus causing two frameshifts yielding a new protein composed of an N-terminal 163 A.A. fragment and a C-terminal 132 A.A. fragment. Because BR-209 may function as an interleukin-1β (IL-1β) binding protein that prevents IL-1β from interacting with the IL-1 receptor, the differences between BR-209 of the Congo versus West African strains of monkeypox may affect virulence [9]. We then used AF2-Batch, the batch-mode AlphaFold2 framework, to predict 3D structural models of all analyzed proteins ( Figure 1B ). In brief, we reimplemented the AlphaFold2 structure-prediction protocol [10] by first decomposing the computation workflow into multiple sequence alignment, end-to-end inference, and structure refinement, then parallelizing the calculations with MPI coding on Slurm based supercomputational infrastructures. Meanwhile, we rewrote the end-to-end structural module along with the TensorFlow backend to avoid multiple compilation of the JAX library. This pipeline enables more than 10000 structure predictions to be made per day on an A100 GPU workstation with ten 50-core CPU nodes, at a speed approximately ten times that of the original AlphaFold2 pipeline. AF2-Batch improved the ability to quickly predict many protein structures, including genome-to-proteome functional studies and structure prediction of systematically mutated proteins. Because of the recent emergence of monkeypox cases, we immediately released the structure models on the website https://www.zelixir.com/Monkeypox/index.html, allowing free use to facilitate further studies (see Data availability).
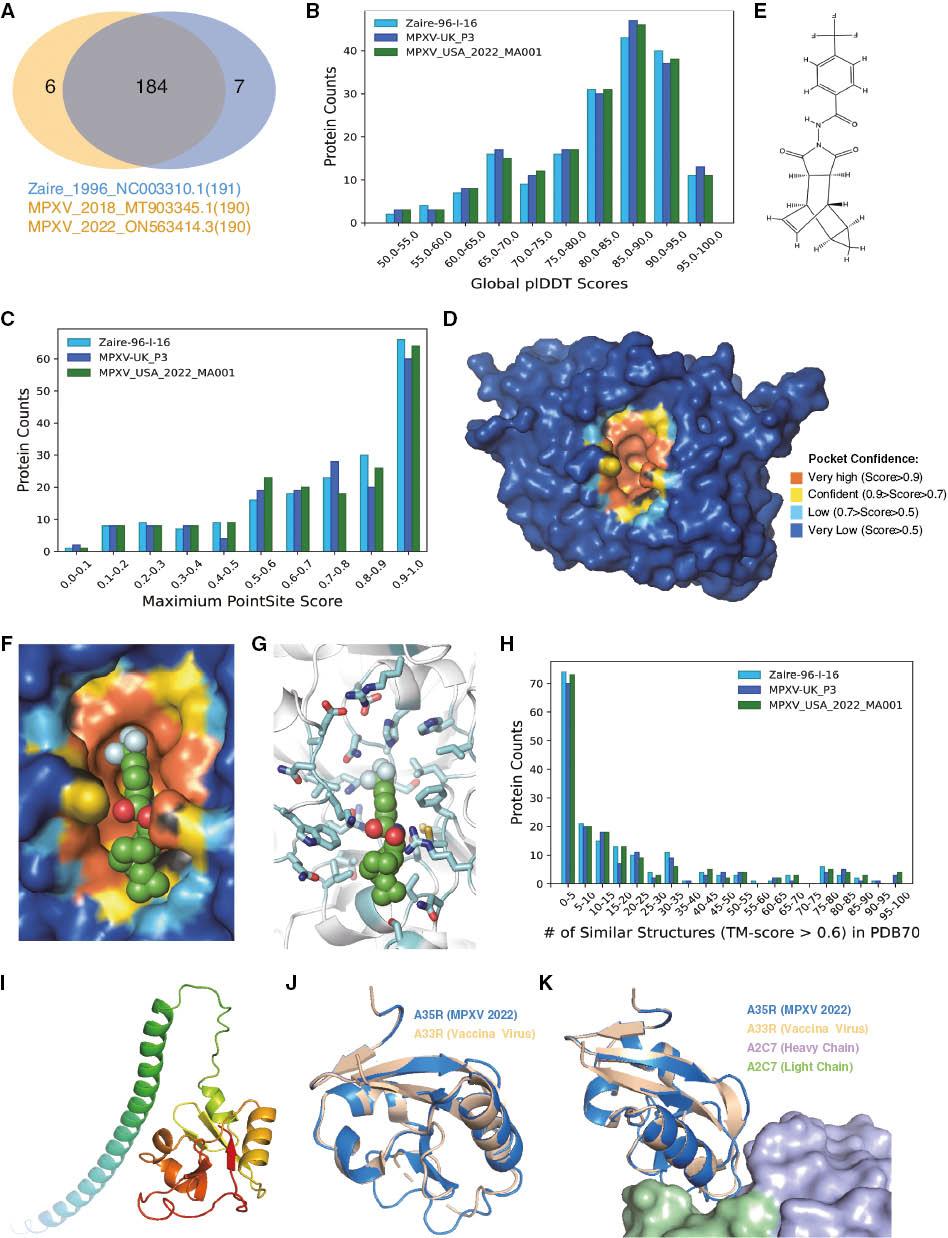
Major differences among 1996 Congo virus strain (Zaire-96-I-16, ID: NC_003310.1), 2018 West African strain (MPXV-UK_P3, ID: MT903345.1), and 2022 West African strain MPXV_USA_2022_MA001, ID: ON563414.3) proteomes. B) plDDT score distribution of the top-ranked structure models of monkeypox virus proteomes. Higher scores indicate higher confidence of the model. C) The PointSite score distribution of the top-ranked structure models of monkeypox virus proteomes. Higher scores indicate greater likelihood of the existence of at least one small-molecule-binding pocket. The panel indicates that at least one-third of the proteins may have small-molecule-binding pockets in the three strains. D) PointSite prediction of the P37 binding pocket. Different colors indicate the confidence of the prediction. The closer the value to 1, the more likely the atom is to be included in the binding region. E) Chemical structure of tecovirimat. F) Tecovirimat fits the pocket of P37 well. Tecovirimat is shown as a green sphere. G) Detailed structure of the P37 pocket with bound tecovirimat. H) Number of similar structures (TM-score > 0.6) and distribution of the top-ranked predicted models of monkeypox virus proteomes. I) Predicted full-length structure of A35R by AF2-batch. Rainbow coloration indicates the main chain from the N terminus to the C terminus. J) Structure alignment of A35R from the monkeypox virus (marine color) 2022 strain and A33R from vaccinia virus (wheat color). K) Recognition of A33R from vaccinia virus and the antibody A2C7. The heavy chain of A2C7 is light blue, and the light chain of A2C7 is pale green.
After completing the protein structure predictions, we implemented the deep PointSite model [11] to annotate the potential binding regions for small molecules on protein surfaces ( Figure 1C ). On the basis of the top-ranking structure model, the models indicated the likelihood of each atom of the protein to compose small-molecule-binding regions. The results have also been released for public use on the website. Here, we chose one of the well-characterized pox proteins, P37, to present the PointSite results. P37 homolog protein, which plays a central role in forming the enveloped viral particle in the smallpox virus, is a validated target for anti-poxviral medication ( Figure 1D ). The closer the value to 1, the more likely the atom is to be part of the binding region. Tecovirimat, the first FDA approved anti-poxviral drug, was approved in 2018 [12] ( Figure 1E ). However, the detailed recognition mechanism of tecovirimat on P37 is unclear. We used PointSite to predict the potential binding site of monkeypox P37. Then we docked tecovirimat in the putative pocket ( Figure 1F ). Tecovirimat fits the pocket well, and the predicted binding energy by AutoDock Vina [13] is approximately -8.0 kcal/mol ( Figure 1G ). This algorithm may aid in rapid selection of proteins with possible small-molecule-binding sites for further drug development targeting other poxviral proteins.
To better study the conserved characteristics of monkeypox proteins, we generated structural alignments against a subset of the Protein Data Bank database (PDB70) [14] for each monkeypox protein, thus obtaining lists of proteins with similar structures ( Figure 1H ). The structure-based protein-alignment-algorithm tool DeepAlign [15] was applied to rank the similarity according to the DeepScore. This function may aid in annotation of unknown proteins’ functions on the basis of structural similarities. Here, we used the protein A35R to present the results ( Figure 1I ). The structural alignment list of A35R, particularly the globular domain, shares high similarity with the A33R of vaccinia virus (PDB ID: 4LQF) ( Figure 1J ) [16]. This structure shows the A33R protein complex with an antibody A2C7 ( Figure 1K ). A33R is a well-known extracellular-enveloped virus (EEV)-specific type II membrane glycoprotein. Because it plays a critical role in efficient EEV formation and facilitates long-range viral spread in hosts, A33 is a potential target for development of neutralizing antibodies targeting EEV. Similarly, A35R of the monkeypox virus is also a target for therapeutic-antibody development to inhibit viral spread.
In summary, we predicted more than 600 structures and added functional annotations of proteins from monkeypox virus proteomes for public use. We provided extensive annotations by using the PointSite algorithm, and labeled the small-molecule-binding regions with high confidence for all 600+ predicted structures. Meanwhile, experimentally determined structures with high similarity to monkeypox proteins were vetted through the structure-alignment algorithm. We hope that our work will accelerate the development of monkeypox vaccines, neutralizing antibodies, and therapeutic drugs.
