
INTRODUCTION
Antibiotic resistance is the natural tendency for bacteria to change under the process of natural selection in such a way that they survive when exposed to the action of previously potent antibiotics. Antibiotic resistance is a rapidly increasing problem, largely as a result of the worldwide misuse and overuse of antibiotics for conditions that do not require them.
Hundreds of thousands of people are dying every year in the world from infections caused by antibiotic-resistant bacteria [1]. In February 2017, the WHO published [2] a list of the most dangerous antibiotic-resistant bacteria – ‘priority pathogens’ that includes 12 families (Table 1). The purpose of this publication was to raise awareness in the world’s scientific community in order to organize and guide the research and development efforts in academia and pharmaceutical industry focused on the search for new antimicrobial agents.
| Priority | Families |
|---|---|
| Critical | 1. Acinetobacter baumannii, carbapenem-resistant 2. Pseudomonas aeruginosa, carbapenem-resistant 3. Enterobacteriaceae, carbapenem-resistant, β-lactamase producing |
| High | 1. Enterococcus faecium, vancomycin-resistant 2. Staphylococcus aureus, methicillin-resistant, vancomycin-intermediate and resistant 3. Helicobacter pylori, clarithromycin-resistant 4. Campylobacter spp., fluoroquinolone-resistant 5. Salmonellae, fluoroquinolone-resistant 6. Neisseria gonorrhoeae, cephalosporin-resistant, fluoroquinolone-resistant |
| Medium | 1. Streptococcus pneumoniae, penicillin-nonsusceptible 2. Haemophilus influenzae, ampicillin-resistant 3. Shigella spp., fluoroquinolone-resistant |
Microorganisms acquire antibiotic resistance through chromosomal DNA mutations and horizontal gene transfer that eventually lead to a change of bacterial proteins. There are several mechanisms of horizontal gene transfer in bacteria – transformation, transduction, and conjugation [3]. Multi-resistant bacteria or ‘superbugs’ have emerged recently due to the horizontal transfer of genetic material that has spread antibiotic resistance.
Recently, scientists from Harvard Medical School (USA) and Technion – Israel Institute of Technology – have designed an experiment visualizing how bacteria acquire resistance to antibiotics [4]. The most dangerous group of multidrug resistant bacteria include E. coli, Klebsiella, Acinetobacter, Serratia, Pseudomonas, Proteus and some other bacteria that cause severe diseases such as bloodstream bacteremia and pneumonia. These infections are easily spread in hospitals and nursing homes and often lead to a lethal outcome.
The goal of this publication is to introduce the challenging and exciting field of the design and discovery of new antibiotics to the wide community of medicinal chemists and scientists interested in drug discovery and development.
The structure of bacteria and types of bacterial resistance to antibiotics
Bacteria being simple organisms contain a well-developed cell structure (Fig. 1). The cell wall provides the structural integrity to bacterial cell and, therefore, it is one of the most important components of the cell envelope. Both Gram-positive and Gram-negative bacteria contain an inner membrane in the form of a phospholipid bilayer. Gram-negative bacteria also have the outer membrane. Gram-positive bacteria do not contain the outer membrane but the cell wall in this type of bacteria is much thicker than that in Gram-negative bacteria. Both Gram-positive and Gram-negative bacteria have DNA, ribosomes and the corresponding enzymes for DNA replication, transcription and translation in order to produce the necessary proteins for the bacterial cell. All the main elements of the bacterial cell envelope as well as organelles and DNA, mRNA, and tRNA molecules and their complexes that are formed in the course of the protein synthesis can be the targets for antibiotics (Fig. 2). The main targets for antibiotics in bacteria are: a) intermediates in the cell wall biosynthesis, b) cell membrane, c) biomolecules and organelles involved in the processes of protein biosynthesis, d) bacterial DNA and RNA, and e) enzymes involved in the biosynthesis of tetrahydrofolate.


Bacteria use several mechanisms for protection from antibiotics that function as mechanisms of resistance. The most common mechanisms of resistance among bacteria are:
d) change in the structure of the intermediates involved in the process of bacterial cell wall synthesis preventing the disruption of this process by antibiotics [13].
Some scientists consider the formation of bacterial biofilms as one of the mechanisms of bacterial resistance, but bacterial biofilms will not be discussed in this review.
Since the efflux of antibiotics that is used by both Gram-positive and Gram-negative bacteria is a general mechanism of resistance to antibiotics that belong to different classes and have quite different molecular structures, it will be briefly discussed in this section. Active efflux is a method that bacteria use for transporting unwanted toxic compounds and antibiotics out of the cell. Efflux pumps are single proteins (Gram-positive bacteria) or a combination of several proteins (Gram-negative bacteria) that are embedded in the bacterial membrane and function like transporters for chemical compounds. There are five distinct types of efflux pumps known to date: MFS (major facilitator class), RND (resistancenodulation-division), SMR (small multidrug resistance class), MATE (multidrug and toxin exclusion), and ABC (ATP-binding cassette). Since the pumping of antibiotics is accomplished often against the concentration gradient, this process requires energy. Most of the efflux pumps – MFS, RND, SMR, and MATE – are powered by the flow of the protons that decreases the proton gradient across the bacterial membrane. The ABC pumps use ATP (adenosine triphosphate) hydrolysis as the energy source.
Mechanisms of action of known antibiotics and the modes of bacterial resistance to them
Natural, semi-synthetic and synthetic antibiotics are successfully used today in clinical practice to fight bacterial infection. Following the generally accepted classification based mostly on molecular structure [16] all marketed antibiotics can be divided into several major classes: β-lactams, glycopeptides and lipoglycopeptides, sulfonamides, aminoglycosides, tetracyclines, macrolides, rifamycines, fluoroquinolones, streptogramines, oxazolidinones, and lipopeptides. An overview of the history of search for antibiotics is given in a paper by Mohr [17]. The mechanisms of action of drugs within each of these antibiotic classes are similar. On the contrary, the mechanisms of action of drugs from different antibiotic classes are significantly different.
The antibiotics structure, mode of action, and mode of bacterial resistance to these antibiotics are presented below. The molecular mechanisms for these processes are discussed.
a. β-Lactams
One of the most widely used classes of antibiotics is β-Lactams (e.g. Fig. 3).

The first antibiotic penicillin was isolated from Penicillium fungi by Alexander Fleming in 1928 [18].
Penicillin belongs to the β-lactam family of antibiotics that have the distinctive structural fragment – four membered β-lactam ring. Cephalosporins (e.g. cefaclor, Fig. 3) also contain β-lactam cycle. The first member of this class of antibiotics – cephalosporin C – was isolated from Cephalosporium acremonium.
There are a number of semisynthetic antibiotics among β-lactams.
Later attempts to find new β-lactam antibiotics led to the development of carbapenems (Fig. 4).

β-Lactam antibiotics act by the inactivation of the transpeptidases that catalyze the last step in peptidoglycan cross-linking. The main component of the cell wall that provides its structural integrity in both Gram-positive and Gram-negative bacteria is peptidoglycan – the crosslinked peptide-sugar copolymer. The cross-linking of the glycan polymer chains that makes the cell wall strong and rigid is accomplished by the formation of peptide strands that connect the glycan chains. The linking of two peptide strands attached to different glycan chains is the last step in the bacterial cell wall synthesis. It is catalyzed by transpeptidases – enzymes that have serine at their active sites. This enzyme attacks the D-alanyl-D-alanyl site of the first peptide strand and forms active acyl-enzyme intermediate with the simultaneous displacement of the terminal alanine (Fig. 5). Then, in Gram-negative bacteria, the amino group of diaminopimelic acid of the second peptide strand reacts with the acyl-enzyme intermediate and the crosslinking reaction is finished by the formation of the peptide bond between the two peptide strands. In Gram-positive bacteria, where diaminopimelic acid in the second peptide strand is replaced by lysine, the connection with the first peptide strand is accomplished through the peptide bridge that can contain several amino acids. For example, in Staphylococcus aureus this peptide bridge contains five glycine amino acids. In this case, the amino group of the last glycine reacts with the acyl-enzyme intermediate to finish the cross-linking process. Penicillins as well as cephalosporins inhibit the enzyme transpeptidase by binding to the active center of enzyme that leads to the formation of the stable acyl enzyme intermediate. As a result of this process, the enzyme cannot catalyze the cross-linking of glycan chains and the process of bacterial wall formation is disrupted as shown in Figs. 6 and 7 [19].



Bacteria developed resistance to penicillin by the production of enzymes β-lactamases [20, 21] and later to cephalosporin drugs by production of cephalosporinases (e. g. class C β-lactamases) [21] and other β-lactamase enzymes known as extended-spectrum β-lactamases (ESBL) [22, 23]. These enzymes cleave the peptide bond in the β-lactam cycle of the corresponding penicillin molecule that leads to the loss of drug activity. (Fig. 8).

The search for methods to overcome bacterial resistance to penicillin antibiotics led to the discovery of a number of β-lactamase enzymes inhibitors like clavulanic acid, sulbactam and tazobactam (Fig. 9) [24, 25]. Since that time, a number of penicillin antibiotics (e.g. ampicillin) were marketed by pharmaceutical companies as mixtures with the corresponding β-lactamase inhibitors. Kuzin et al. [25] presented the X-ray crystallography data for the SHV-1 class A β-lactamase-tazobactam complex and discussed the mechanism of this enzyme inhibition by tazobactam. Mechanism of action of avibactam (Fig. 9), the effective inhibitor of class A, C and some class D β-lactamases and the first non-β-lactam β-lactamase inhibitor, is discussed in recent publications [26, 27].

The combination of tazobactam with the new antibiotic ceftolozane (Fig. 10) that belongs to the 5th generation of cephalosporins is marketed as Zerbaxa. This antibiotic shows high potency against several drug resistant strains [28, 29].

The strategies to discover new β-lactam antibiotics and the development of bacterial resistance to them are reviewed by Llarrull et al. [30]. Unfortunately, the known β-lactamase enzymes inhibitors that are active against class A, C, and D β-lactamases cannot deactivate class B metallo-β-lactamases. These enzymes, containing Zn in their active site, utilize a different mechanism to cleave the β-lactam cycle of antibiotics. Monobactams [31] (Fig. 11) are the only group of β-lactam antibiotics that can effectively inhibit those enzymes [32, 33]. Thus, the results published by Marshall et al. [34] suggest the combination of ceftazidime-avibactam and aztreonam could be used to overcome resistance of bacteria producing metallo-β-lactamases.

In addition, known β-lactamase enzyme inhibitors are not active enough against the extended-spectrum cephalosporinases [35, 36]. Today, the search for the potent inhibitors of this class of enzymes is an area of active exploration [26, 37]. On the other hand, the search for the new types of β-lactamases inhibitors continues. Thus, Venugopal et al. [38] published interesting results on using peptidyl boronate analogues as broad-spectrum β-lactamase inhibitors.
Another approach to combatting β-lactamase resistance is to block one of the key steps in the biosynthesis of these enzymes that was demonstrated by Bouquet et al. for AmpC β-lactamase [39]. Recently discovered 2-aminobenzimidazoles were found to significantly reduce the bacterial resistance to a number of β-lactam antibiotics [40]. It is believed that these compounds have a different mechanism of action from the β-lactamase inhibitors.
Expression by the bacteria of the mutant enzyme transpeptidase (mutant penicillin-binding protein – PBP2 enzyme) that has a reaction center practically inaccessible to the β-lactam antibiotics but, on the other hand, perfectly capable of catalyzing the crosslinking reaction in the course of peptidoglycan synthesis, serves as another way of bacterial resistance to β-lactam antibiotics [41]. It was discovered that in order to catalyze the crosslinking of peptidoglycan the active center of the enzyme should undergo conformational change. This conformational change is probably triggered by the interaction of the corresponding peptidoglycan fragments with the allosteric site of this enzyme [41]. Then, it was shown that some β-lactams like ceftaroline are able to interact with the allosteric site of the enzyme leading to the conformational change – opening of its active site – that enables the second molecule of ceftaroline to react with the enzyme active site, thereby finishing the enzyme inactivation [41, 42]. Fishovitz et al. reported that the triggering of the allosteric cite of PBP2 enzyme by one β-lactam antibiotic enables another β-lactam drug to interact with the active cite of that protein. Therefore, they report that there is synergy in the action of ceftaroline and some other β-lactams like oxacillin, imipenem, and meropenem against the MRSA bacterial strains [43].
Other mechanisms of antibiotic resistance in Gramnegative bacteria involve structural and/or functional changes in porin superfamily transporters that are imbedded in the bacterial outer membrane. These changes strongly affect the rate of antibiotics permeation though the bacterial outer membrane. It was shown that mutations of porins or switching to the expression of the other type porins with a different pore size or different electrostatic interactions in the porin eyelet region lead to a decrease in the permeability of β-lactams in Gram-negative bacteria that serves as the reason for bacterial resistance to these antibiotics [44, 45].
b. Glycopeptides and Lipoglycopeptides
Another class of antibiotics that act by preventing the biosynthesis of the bacterial cell wall is glycopeptides (e. g. Fig. 12) and their semisynthetic derivatives – lipoglycopeptides (e. g. Fig. 17).

Both vancomycin and teicoplanin are natural antibiotics produced by microorganisms. Vancomycin was isolated from the soil bacteria Amycolatopsis orientalis. Teicoplanin is a mixture of natural antibiotics isolated from bacteria Actinoplanes teichomyceticus. Glycopeptides act as inhibitors of the peptidoglycan synthesis in bacterial cell wall. Vancomycin binds to the D-alanyl-D-alanyl end fragment in the peptide strands of the peptidoglycan intermediate and prevents the cross-linking of the peptide polymer chains (Fig. 13).

The bacterial strains resistant to vancomycin emerged as a response to the action of this antibiotic. These new bacterial strains have cell walls with a different structure of peptide chains terminus – D-alanyl-D-lactate – instead of D-alanyl-D-alanyl. Since vancomycin forms 5 hydrogen bonds with a D-alanyl-D-alanyl fragment and only 4 hydrogen bonds with D-alanyl-D-lactate fragment (Fig. 14) its activity decreases 1,000-fold against these vancomycin-resistant bacteria. Xie et al. [46] reported the synthesis of vancomycin aglycon (Fig. 15) specially designed to interact with both D-alanyl-D-alanyl and modified D-alanyl-D-lactate fragments in peptide strands. This compound showed high potency against vancomycin-resistant bacteria.


Silverman et al. [47] described the synthesis of vancomycin dimers and showed that these compounds have increased potency against methicillin-resistant Staphylococcus aureus and some vancomycin resistant strains.
An interesting approach to improve vancomycin activity against resistant bacteria was used by Okano et al. [48]. They changed one functional group and introduced 2 new functional groups in the molecule of vancomycin (Fig. 16). These new functional groups enable the new drug to act as an antibacterial agent according to two different mechanisms. As a result, the new vancomycin drug molecule has three possible different mechanisms of action and showed high activity against vancomycinresistant bacteria.

Kang et al. [49] showed that combinations of vancomycin with non-β-lactam antibiotics like ciprofloxacin and gentamicin give a synergetic effect against bacteria with reduced susceptibility to vancomycin. Yarlagadda et al. [50] showed that lipophilic vancomycin aglycon dimer is 300 times more active compared to vancomycin against some vancomycin resistant bacteria. They also reported that vancomycin-sugar derivatives, especially lipophilic vancomycin-sugar analog, exhibit significantly higher activity against the vancomycin resistant strains than vancomycin itself [51].
In attempt to improve the activity of vancomycin a number of its semisynthetic derivatives has been isolated and studied [52]. As a result of these research efforts, three new drugs – dalbavancin, telavancin and oritavancin, that belong to the subclass of lipoglycopeptides (Fig. 17) were introduced to the market. They have certain advantages as compared with vancomycin. Dalbavancin requires fewer frequent dosing because it has much longer half-life. The lipophilic fragments in the structures of both telavancin and oritavancin enable these drugs to have a second mechanism of action – to induce the membrane depolarization in bacterial cell. Due to this dual-action mechanism, both antibiotics show excellent antibacterial activity. Telavancin also has significantly improved ADME (adsorption, distribution, metabolism and excretion) profile [52, 53].

c. Sulfonamides
Sulfonamides represent another important class of antibiotics that inhibit the synthesis of the folic acid metabolite tetrahydrofolate. This compound is an important intermediate in different biosynthetic reactions and also plays an important part in the cell division process. Sulfanilamide and sulfamethoxazole are the main examples of sulfonamides (Fig. 18). Sulfanilamide is a synthetic antibiotic the structure of which is close to that of p-Aminobenzoic acid (PABA) (Fig. 19).


Sulfonamide antibiotics mimic p-aminobenzoic acid in the process of tetrahydrofolate synthesis (Fig. 20) and are potent against both Gram-negative and Gram-positive bacteria. By interacting with enzyme Dihydropteroate Synthase (DHPS) this class of antibiotics prevents the formation of intermediate I in the synthesis of tetrahydrofolate (Fig. 20) [54, 55].

Resistance to sulfonamide antibiotics developed because the small fraction of bacteria can use very small concentrations of folic acid provided by the host organism. Since bacteria reproduce fast, even a small number of surviving bacterial cells results in the spread of infectious disease in humans. Another mechanism of bacterial resistance to sulfonamides [56] involves certain mutations in enzyme DHPS that make it practically insensitive to these drugs. At the same time, the mutant enzyme preserves the ability to bind the PABA that is structurally very similar to sulfonamides.
Search for the inhibitors of the enzyme dihydrofolate reductase (DHFR) that catalyzes the conversion of dihydrofolic acid into tetrahydrofolate, led to the discovery of diaminopyrimidines – the second class of synthetic drugs that disrupt the tetrahydrofolate synthesis in bacteria. These compounds (e.g. trimethoprim, Fig. 21) prevent the reduction step in the synthesis of tetrahydrofolate.

Trimethoprim inhibits the reduction of dihydrofolic acid to tetrahydrofolic acid by binding to the enzyme dihydrofolate reductase (DHFR). Therefore, the combination of sulfonamide antibiotics with diaminopyrimidines was successfully used in fighting both Gram-positive and Gram-negative bacteria. Resistance to trimethoprim in some pathogenic bacteria developed as a result of the single amino acid Ile100Leu substitution in dihydrofolate reductase (DHFR) [57]. Recently, Nepka et al. [58] showed that the combination of sulfamethoxazole-trimethoprim with colistin could be effectively used for fighting carbapenem-resistant bacterial strains. Bavadi et al. describe the synthesis of the series of new dihydropyrrol-2-one compounds containing sulfonamide groups. Most of these compounds showed antibacterial activity against P. aeruginosa and S. epidermidis and were more active against the P. aeruginosa than the combination of trimethoprim-sulfamethoxazole [59].
d. Aminoglycosides
Another group of antibiotics (streptogramins, macrolides, oxazolidinones, tetracyclines and aminoglycosides) kills bacteria by disrupting its protein synthesis. The targets for these drugs are both large 50S and small 30S ribosomal subunits. One of the antibiotic classes that target the bacterial ribosome is aminoglycosides (e.g. streptomycin and kanamycin Fig. 22).

Aminoglycosides are natural antibiotics. Streptomycin – the first member of this class of antibiotics – was isolated from the Gram-positive bacteria Streptomyces griseus. Streptomycin inhibits the protein synthesis in bacteria by binding to the 30S subunit of the bacterial ribosome. That leads to codon misreading over the course of the translation process, resulting in the synthesis of proteins with wrong structures and eventually to bacterial death. Interaction of neomycin with 50S ribosomal subunit is shown below (Fig. 23) [60].
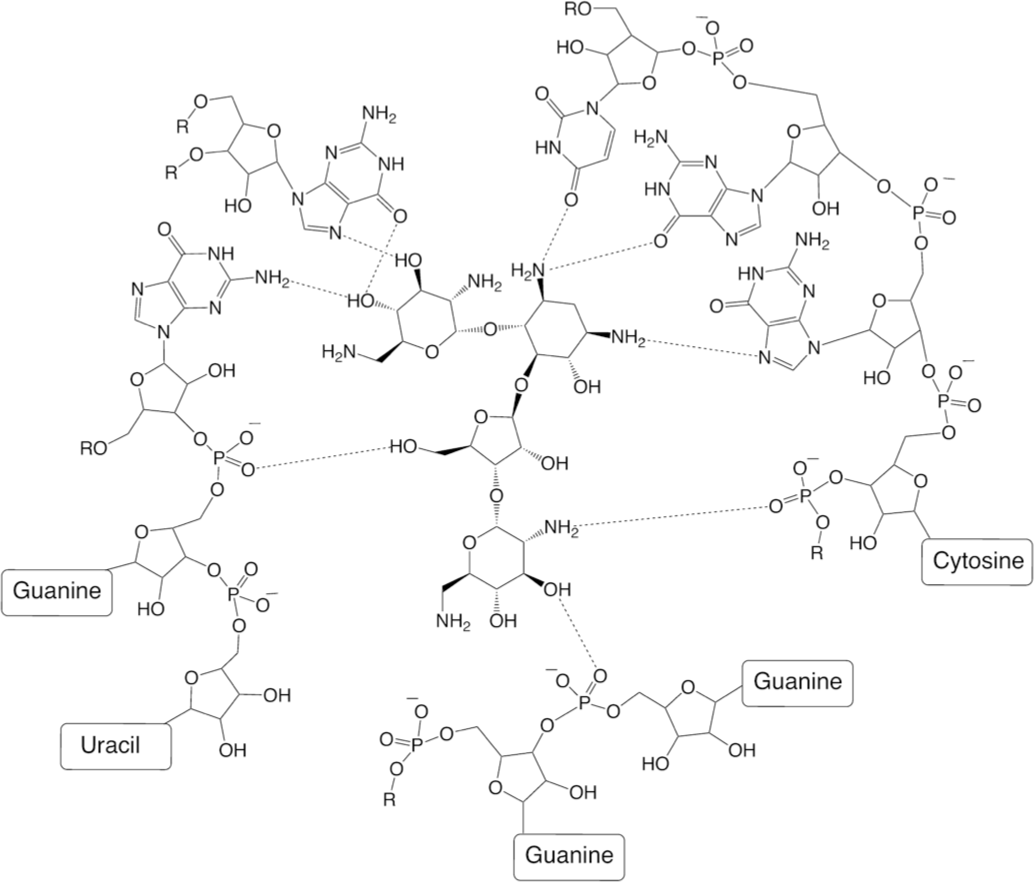
Streptomycin-resistant bacterial strains (both Grampositive and Gram-negative) emerged after the introduction of this antibiotic and its analogues. Action of several bacterial enzymes such as acetylase, adenylase, and phosphorylase leads to the deactivation of aminoglycoside antibiotics [7, 8] (Fig. 24). Enzymatic acetylation, phosphorylation, and adenylylation produce modified drug molecules that have lost their antibacterial activity.

In order to overcome bacterial resistance, new semisynthetic aminoglycoside antibiotic amikacin (Fig. 25) was designed. This compound is stable to enzymatic deactivation. The amikacin binds to the A-site of 16S rRNA that disrupts the protein synthesis.

In this molecule, the L-hydroxyaminobutyramide moiety attached to the central ring fragment protects the drug from attacks by certain bacterial antibiotic inactivating enzymes. Another semisynthetic drug candidate that is stable to enzymatic acylation, phosphorylation, and adenylylation – plazomicin (Fig. 26) – is now in the development stage.

e. Tetracyclines
Another class of broad-spectrum antibiotics that target the 30S ribosomal subunit is tetracyclines (Fig. 27).

Tetracyclines are natural products. The first member of this class of antibiotics – chlorotetracycline – was isolated from the bacteria Streptomyces aureofaciens. There are also a number of semisynthetic tetracycline drugs. In the review by Liu et al. practical methods of the synthesis of tetracyclines are summarized including an example of the successful synthesis of a new potent antibiotic eravacycline that is currently in clinical trials [61].
Tetracyclines are broad-spectrum antibiotics active against both Gram-negative and Gram-positive bacteria. Tetracyclines bind to the 30S subunit of the ribosome in bacterial cell (an example: tigecycline, Fig. 28) [62] and disrupt the translation (synthesis of proteins) process by preventing aminoacyl-tRNA from binding to the corresponding site of mRNA – ribosome complex.

Since this class of antibiotics was used extensively, the bacterial resistance emerged without delay. Three major mechanisms of resistance to tetracyclines have been recognized to date: efflux of the drug molecules from the bacterial cell by the efflux pumps, enzymatic modification of tetracycline molecule leading to its inactivation and ribosomal protection [63]. The latter involve the synthesis of ribosomal protection proteins (RPPs) (e.g. Tet(O) and Tet(M)) that significantly reduce the binding of tetracycline antibiotics to the ribosome subunit or displace them from the binding site. Ribosomal protection is considered to be one of the two major mechanisms of bacterial resistance against tetracyclines [64]. This approach is used by both Gram-positive and Gram-negative bacteria.
One of the efficient ways to fight the drug resistant strains of bacteria is the chemical modification of existing antibiotics. By using this approach, new representatives of this class of drugs efficient against the resistant strains were discovered [65, 66] (Fig. 29).

Another major mechanism of tetracyclines resistance in bacteria is drug efflux from bacterial cell. It was proven that membrane-associated proteins (e.g. Tet(A), Tet(B), Tet(C)) are responsible for the efflux of tetracyclines from bacteria [63]. Most of them belong to the major facilitator superfamily (MFS) of efflux transporters [9, 10, 67] though some of them (e.g. EfrAB [9, 68]) belong to the ATP-binding cassette (ABC) family of efflux pumps. Thus, Tet proteins transport tetracycline molecules coordinated with the divalent cations (Ca2+ or Mg2+) through the bacterial membrane in exchange for the protons [67, 10]. Nelson et al. [69] demonstrated that cyclopentylthiotetracycline (13-CPTC) is an efficient inhibitor of Tet(B) protein. The uptake of tetracycline into the resistant bacteria was significantly increased in the presence of 13-CPTC (Fig. 30) as a result of bacterial efflux pumps blockage. Therefore, the simultaneous administration of 13-CPTC with tetracycline can restore the activity of this antibiotic against the resistant bacteria.

A new antibiotic fluorocycline TP-271 (Fig. 31), described recently by Grossman et al. [70], demonstrates high in vitro potency against multidrug-resistant bacteria. These results once more show that changing the antibiotic’s structure is a promising approach to combat bacterial resistance.

The third – less prevalent – mechanism of resistance is the chemical modification of tetracyclines studied by Speer et al. [71]. They found that tetracycline is chemically modified in bacteria that carry the *Tcr gene in aerobic conditions. The structures of modified tetracycline products were not determined. The impact of tetracyclines coordination with metals on antimicrobial activity was reviewed by Guerra et al. [72].
f. Streptogramins
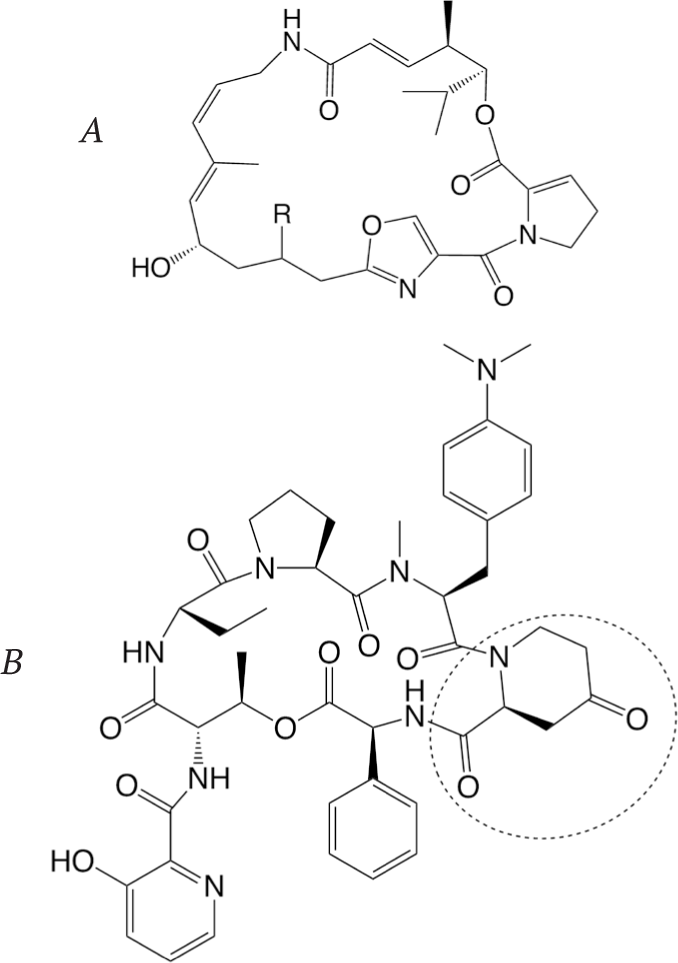
One of the antibiotic classes that target the large 50S ribosomal subunit is streptogramins (e.g. dalfopristin (streptogramin A) and quinupristin (streptogramin B), Fig. 32).

Both dalfopristin and quinupristin are semisynthetic modifications of the natural streptogramin antibiotics produced by various Streptomyces bacteria. Streptogramins inhibit the bacterial protein synthesis by binding to the 50S ribosomal subunit. Group A streptogramins (dalfopristin) bind in the peptidyltransferase center (PTC) and interfere with binding of aminoacyltRNAs, which prevents the formation of a peptide bond during the elongation step. Streptogramin B components (quinupristin) bind to the proximal end of the exit tunnel resulting in the release of the small incomplete oligopeptide chains [73, 74]. Each compound shows moderate activity, whereas the combination of both drugs demonstrates a synergetic effect. The binding of dalfopristin results in a specific conformational change of the 50S ribosomal subunit that significantly facilitates the consequent binding of quinupristin. As a result, administration of the dalfopristin and quinupristin mixture (marketed as Synercid) produces much higher antibacterial activity.
Bacteria have developed several mechanisms of resistance to these antibiotics including active efflux from the bacterial cell, 23S rRNA methylation, and enzymatic deactivation. The acetylation of the OH group in dalfopristin and its analogues catalyzed by O-acetyltransferases (e.g. virginiamycin acetyltransferase) results in the formation of O-acetyl products that do not have the binding affinity to 50S ribosome subunit [73]. Enzymatic deactivation of quinupristin and its structural homologues proceeds by lactone opening catalyzed by virginiamycin B lyase (Vgb) resulting in the formation of linear products that have lost their antibacterial activity [75].
This development accelerated the search for new streptogramins active against the resistant strains. The main efforts to modify the structure of type A streptogramins were concentrated on the introduction of different substituents R in position 16 of 16-desoxopristinamycin II (Fig. 33). On the other hand, the substitution of the 4-oxo2-piperidinecarboxylic acid fragment in pristinamycin I (Fig. 33) by other heterocyclic amino acids led to the discovery of several new active compounds [73]. An interesting approach to the modification of the streptogramin structure was demonstrated by Mukhtar et al. [76]. Using synthetic and enzymatic methods, they prepared a series of chimeric compounds containing structural elements of streptogramins B and antibiotic tyrocidine. These compounds showed a broad range of activity against Grampositive bacteria. It is believed that their mechanism of action is different from that of the parent antibiotics.
In a recent publication by Li et al., a new practical scalable approach to the synthesis of streptogramin antibiotics is described. This method can lead to the discovery of new potent drugs that will overcome the limitations of known streptogramins [77].
g. Macrolides
Another class of antibiotics that target the large ribosomal subunit is macrolides (e.g. azithromycin and erythromycin, Fig. 34).

Erythromycin is a natural compound isolated from the bacteria Saccharopolyspora erythraea. Azithromycin is one of semisynthetic analogues of erythromycin. Erythromycin binds close to the peptidyl transferase center (PTC) in the 50S ribosomal subunit at the end of the polypeptide exit tunnel. It affects the polypeptide chain elongation step and consequently disrupts the whole process of protein biosynthesis [78]. The mechanism of action of these antibiotics is discussed by Gaynor et al. [79].
During last two decades, we witnessed the emergence of bacterial resistance to macrolides and particularly to erythromycin [80]. There are three known mechanisms of resistance. The primary mechanism involves post-transcriptional methylation of the 23S ribosomal RNA that prevents the antibiotic from binding to bacterial ribosome [79]. The other mechanism of resistance is based on the action of efflux pumps that exert antibiotics from the bacterial cell [81]. One more possible mechanism involves the ribosomal synthesis of small peptides (4-6 amino acids) that interact with the drug molecule bound to the 50S subunit of the ribosome and promote its release from the active site [79]. In order to fight bacterial resistance, the second (clarithromycin) and then the third (telithromycin) generation of macrolides were developed (Fig. 35). The latter has a significantly improved affinity to both the native and mutant 50S ribosome subunits, although the new mutations that lead to this antibiotic resistance have already been reported [81].

Summary of practical methods of synthesis of new macrolide antibiotics is presented by Seiple et al. [82].
h. Oxazolidinones
Oxazolidinones (e.g. linezolid and tedizolid, Fig. 36) are the only completely synthetic class of antibiotics that target bacterial ribosome.
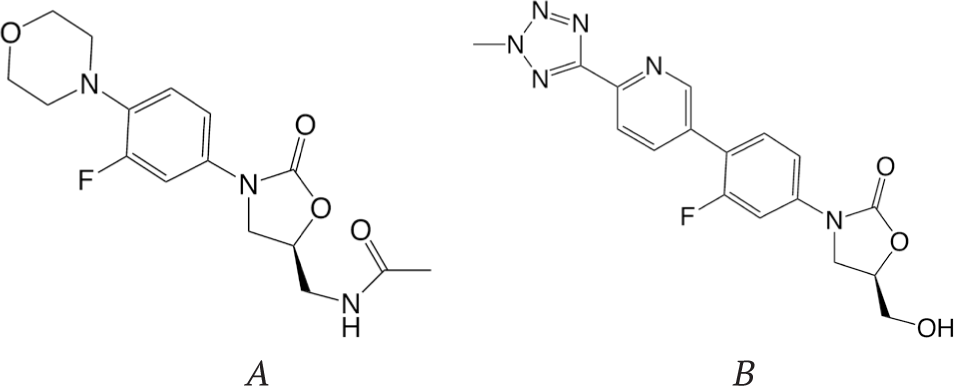
The distinctive structural fragment of this class of antibiotics is the five-membered oxazolidinone ring. This class of compounds was first investigated at DuPont and then at Pharmacia [73]. Linezolid and other oxazolidinones bind to the 50S ribosomal subunit and inhibit the process of protein synthesis. The possible mechanism of action of oxazolidinones is binding at the P site of the 50S ribosomal subunit and inhibition of the initiation step of translation [73]. Resistance to oxazolidinones occurs by mutations in the binding site of these antibiotics at the 50S subunit. The second generation of oxazolidinones – antibiotic tedizolid, which was approved for use in humans in 2014, – has improved potency and broader antimicrobial activity. A series of new oxazolidinones containing fused heterocyclic rings were synthesized and showed higher activity than linezolid against some Gram-positive and Gram-negative bacteria [83].
Fujisaki et al. report the synthesis of novel oxazolidinone dimers that showed significant activity against both Gram-negative and Gram-positive bacteria [84].
Interesting approach in search for new antibiotics was demonstrated by Gordeev et al. [85] and Hubschwerlen et al. [86, 87]. They synthesized new antibiotics by linking ciprofloxacin (fluoroquinolone) and oxazolidinone fragments together. The best representative of this class of compounds (Fig. 37) showed broad antibacterial activity including activity against fluoroquinoloneand linezolidresistant bacteria.

i. Flouroquinolones
Another important group of antibiotics selectively targets certain steps of bacterial DNA replication and transcription. Synthetic antibiotics that belong to the flouroquinolone class inhibit DNA replication while the natural and semisynthetic rifamycins disrupt the transcription process.
Fluoroquinolones (e.g. ciprofloxacin and norfloxacin, Fig. 38) are considered to be the most successful class of synthetic antibiotics.

Both ciprofloxacin and norfloxacin are synthetic antibiotics containing the central quinolone fused ring system.
Fruoroquinolones are broad-spectrum antibiotics. They interact with enzymes DNA gyrase and topoisomerase IV that leads to the distraction of the DNA replication process in the bacterial cell and also prevents the bacterial DNA repair and disrupts the cell division process. In the course of the DNA replication process, the double stranded DNA is unwound by the DNA helicase enzyme. That leads to an under wound DNA sector (negative supercoils) on one side of the replication fork and super wound DNA sector (positive supercoils) on the other side. The main role of DNA gyrase is to relieve the DNA supercoiling strand. This is achieved by DNA double strand cleavage that leads to the relief of both negative and positive supercoiling, and then the successful resealing of the DNA double strand break. Flouroquinolones interact with the double-strand cleaved DNA-enzyme complexes, stabilize them, and prevent DNA double-strand resealing [88]. The role of enzyme topoisomerase IV is to release the two daughter chromosomal circles after the replication step that is essential in the process of cell division. This is achieved by cutting the DNA double –strand of one of the chromosomes, moving the cut site out of the second chromosome and the consequent resealing of the cut. Fluoroquinolones interact with the complex formed by enzyme topoisomerase IV with DNA double-strand cut and stabilize it (Fig. 39) [88]. That, in turn, prevents the sealing of the double-strand DNA cuts and leads to DNA breakage and eventually to bacterial cell death. In spite of the wide use of fluoroquinolones in medical practice, the detailed molecular mechanism of action of these drugs was discovered quite recently.

Bacterial resistance to this class of antibiotics emerged as a result of their widespread use for the treatment of infections in humans. In many cases, these antibiotics were misused for the treatment of infections that could be treated with other narrow spectrum antimicrobials and even for viral infections.
There are several known mechanisms of resistance to quinolones in Staphylococcus aureus. One of them – plasmid-mediated – is an active efflux of these antibiotics from the bacterial cell by drug pumps. It has been shown [89-92] that the NorA efflux pumps are responsible for the decreasing of the concentration of some (hydrophilic) fluoroquinolone drugs in the bacterial cell and consequently for the loss of activity of these antibiotics in Staphylococcus aureus. According to the recent publication by Tintino et al. [93], the tannic acid is an efficient inhibitor of the NorA efflux pumps in this bacterial strain. Thus, they showed that the administration of norfloxacin together with tannic acid (Fig. 40C) leads to a significant decrease in the minimum inhibitory concentration (MIC) for this antibiotic. It was also shown that reserpine is an efficient inhibitor of the NorA efflux pumps [94]. Other possible inhibitors for these efflux pumps include prochlorperazine, paroxetine, verapamil, and omeprazole (Fig. 40 A, B, D, E, correspondingly) [9], although in order to achieve significant inhibition a relatively high concentration of these compounds is necessary.

Since active efflux is one of the major mechanisms of resistance [95], the search for the new efflux pumps inhibitors continues. Coelho et al. [96] showed that nerol (Fig. 41) acting as a drug pump inhibitor can significantly increase the activity of norfloxacin against the Staphylococcus aureus strain. According to Felicetti et al. [97] 2-(3,4-dimethoxyphenyl)quinoline (Fig. 41) is an active NorA pumps inhibitor and can be successfully used in combination with ciprofloxacin.

Other plasmid-mediated resistance mechanisms include the bacterial production of pentapeptide repeat proteins that prevent the drugs from binding to DNA gyrase and action of bacterial enzyme aminoglycoside acetyltransferase AAC (6’)-Ib-cr leading to the drug inactivation [98, 99]. Two codon changes in aac (6’)-Ib gene directing two substitutions Trp102Arg and Asp179Tyr lead to structural changes in aminoglycoside acetyltransferase that enable this enzyme to acetylate the ciprofloxacin. After the acylation of NH-group of the piperazinyl fragment (Fig. 42), the drug shows practically no antibacterial activity.

Obviously, this mechanism of resistance affects only those fluoroquinolones that have a piperazinyl ring like ciprofloxacin, norfloxacin, etc. Other representatives of this class of antibiotics like rufloxacin, perfloxacin, and fleroxacin that have an N-methylpiperazinyl fragment (Fig. 43) are unaffected by bacterial aminoglycoside acetyltransferase AAC(6’)-Ib-cr [99].

In order to overcome the bacterial resistance, a search for the new fluoroquinolone antibiotics is ongoing. According to Abu-Qatouseh et al. [100], a number of 8-nitrofluоroquinolones are active against some strains of Helicobacter pylori. Some of these compounds have shown synergetic effects when combined with metronidazole. Compounds from other chemical classes are also considered as topoisomerase IV inhibitors. A new gyrase inhibitor spiropyrimidinetrione AZD0914 (Fig. 44) was developed by AstraZeneca [101]. Another new lead compound for gyrase inhibition (Fig. 44, compound 2) was reported by Chan et al. [102].

j. Rifamycins
Rifamycins represent the natural and semisynthetic class of antibiotics that target RNA polymerase. Natural rifamycins are produced by Amycolatopsis rifamycinica (previously known as Amycolatopsis mediterranei) although they can also be synthesized in the laboratory. Rifampicin, rifapentine, and rifabutin are examples of semisynthetic rifamycins (Fig. 45).

Semisynthetic drugs of this class (e.g. rifampicin) have rather broad-spectrum antibiotic activity. Rifamycins bind to the RNA polymerase in the bacterial cell and distract the transcription process – DNA dependent synthesis of mRNA [103]. Interaction of the drug molecule with the enzyme leads to the formation of a complex that prevents the elongation of the mRNA oligonucleotide chain. These drugs are active against the Gram-positive and some strains of Gram-negative bacteria. Mutations in the bacterial RNA polymerase, which can develop very quickly, lead to the strong bacterial resistance to these antibiotics. Rifamycin producer Amycolatopsis rifamycinica uses this approach for self-protection. The effect of different mutations in this enzyme on antibiotic activity of rifamycins was discussed recently [104, 105]. General overview of this class of antibiotics with a focus on zwitterionic compounds is given in [106]. Since these antibiotics are used to treat tuberculosis, there is an ongoing search for new active compounds. Thus, Bujnowski et al. [107] reviewed the development of new methods for the synthesis of novel rifamycins, whereas Czerwonka et al. [108] described a new series of these antibiotics containing L-amino acid esters and showed that some of them have antibacterial activity comparable to rifampicin.
k. Lipopeptides
Lipopeptides are the unique class of antibiotics that target the bacterial membrane. Bacterial membranes have significantly higher content of anionic phospholipids than membranes of eukaryotic cells. That allows for the selective targeting of bacterial membranes by antibiotics. There are several known active lipopeptide antimicrobials and at least three of them –polymyxin B, polymyxin E (colistin) (Fig. 46) [109], and daptomycin (Fig. 47) [109] – are or have been in clinical practice.


Polymyxins are natural antibiotics produced by different strains of Paenibacillus polymyxa. Daptomycin is also a natural compound isolated from bacteria Streptomyces roseosporus, although recently a synthetic method for this compound was developed. Daptomycin as well as polymyxins is synthesized in bacteria by complexes of nonribosomal peptide synthetase enzymes. Polymyxins are a mixture of several compounds; the major components are shown in Fig. 46.
The action of these antibiotics leads to a drastic change in bacterial membrane permeability and eventually causes bacterial cell death. Thus, it is believed that daptomycin initially interacts with the negatively charged bacterial membrane by means of coordination through the Ca2+ ions, then its lipophilic ‘tail’ is inserted in the membrane bilayer and the consequent interaction with several other daptomycin molecules leads to oligomerization, and finally ion channel formation (Fig. 48). The formation of an ion channel leads to potassium ions efflux from the bacterial cell, membrane depolarization, and eventually bacterial cell death [111]. Resistance to daptomycin in several bacterial strains has been reported. The mechanism of resistance is related to the changes in bacterial membrane – from significant decrease to the absence of anionic phospholipids in the membrane structure [14]. These changes are connected with the loss of cdsA activity that encodes the cdsA enzyme involved in synthesis of membrane anionic lipids [15]. In the absence of a negative charge on the bacterial membrane daptomycin cannot interact with it through the Ca 2+ ions and thereby bacteria become resistant to this antibiotic.

Step I – binding and insertion, step II – oligomerization, step III – channel formation and ion efflux.
The recent developments in search for new macrocyclic polypeptide antibiotics including glycopeptides and lipopeptides was recently reviewed by Luther et al. [112]. In general, antimicrobial peptides (AMPs) are getting more and more attention as novel promising therapeutic agents against antibiotic resistant bacteria [113-115].
CONCLUSION
In conclusion, we can summarize that the general methods to overcome bacterial resistance are:
Discovery of new antibiotics that are active against drug resistant bacteria due to:
Simultaneous administration of two antibiotics with a synergetic effect.
New antibiotics that are in the late stages of clinical development were recently reviewed by Fernandes et al. [125].
It should be mentioned that there are a number of new approaches in the development of antibiotics active against the drug resistant bacteria e.g. development of pleuromutilins, quorum-sensing inhibitors, FabI-inhibitors, peptidomimetics, inhibitors of bacterial virulence etc.
These are definitely very important and promising areas of research, although no approved antibiotics for use in humans (except retapamulin approved for topical use) have been discovered so far.
The rapid spread of antibiotic resistance in bacteria makes it necessary to intensify the development of new antibiotics and expand the search for new methods to fight bacterial resistance. Over the course of these research efforts, scientists are gaining a better understanding of the mechanisms of antibiotic action, the mechanisms of bacterial resistance, and the key biochemical processes of the bacterial life cycle. On the other hand, new targets for antibiotics in bacteria are being discovered [126].