- Record: found
- Abstract: found
- Article: found
New Therapeutic Options for the Treatment of Sickle Cell Disease
Read this article at
Abstract
Sickle cell disease (SCD; ORPHA232; OMIM # 603903) is a chronic and invalidating disorder distributed worldwide, with high morbidity and mortality. Given the disease complexity and the multiplicity of pathophysiological targets, development of new therapeutic options is critical, despite the positive effects of hydroxyurea (HU), for many years the only approved drug for SCD.
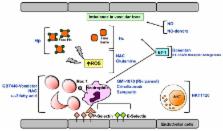
New therapeutic strategies might be divided into (1) pathophysiology-related novel therapies and (2) innovations in curative therapeutic options such as hematopoietic stem cell transplantation and gene therapy. The pathophysiology related novel therapies are: a) Agents which reduce sickling or prevent sickle red cell dehydration; b) Agents targeting SCD vasculopathy and sickle cell-endothelial adhesive events; c) Anti-oxidant agents.
This review highlights new therapeutic strategies in SCD and discusses future developments, research implications, and possible innovative clinical trials.
Related collections
Most cited references106
- Record: found
- Abstract: found
- Article: not found
Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease.
- Record: found
- Abstract: not found
- Article: not found
Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease
- Record: found
- Abstract: found
- Article: not found