- Record: found
- Abstract: found
- Article: found
LKB1 suppresses androgen synthesis in a mouse model of hyperandrogenism via IGF‐1 signaling
Read this article at
Abstract
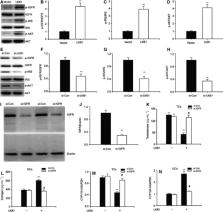
Polycystic ovary syndrome ( PCOS) is a major cause of anovulatory sterility in women, and most PCOS patients exhibit hyperandrogenism ( HA). Liver kinase b1 ( LKB1) is a tumor suppressor that has recently been reported to be involved in PCOS. However, the mechanism by which LKB1 affects HA has not previously been elucidated. We report here that ovarian LKB1 levels are significantly decreased in a female mouse model of HA. Moreover, we report that LKB1 expression is inhibited by elevated androgens via activation of androgen receptors. In addition, LKB1 treatment was observed to suppress androgen synthesis in theca cells and promote estrogen production in granulosa cells by regulating steroidogenic enzyme expression. As expected, LKB1 knockdown inhibited estrogen levels and enhanced androgen levels, and LKB1‐transgenic mice were protected against HA. The effect of LKB1 appears to be mediated via IGF‐1 signaling. In summary, we describe here a key role for LKB1 in controlling sex hormone levels.
Related collections
Most cited references36
- Record: found
- Abstract: found
- Article: not found
Development of polycystic ovary syndrome: involvement of genetic and environmental factors.

- Record: found
- Abstract: found
- Article: found
FOXO3 Is a Glucocorticoid Receptor Target and Regulates LKB1 and Its Own Expression Based on Cellular AMP Levels via a Positive Autoregulatory Loop
- Record: found
- Abstract: found
- Article: not found