- Record: found
- Abstract: found
- Article: found
Two Rare Human Mitofusin 2 Mutations Alter Mitochondrial Dynamics and Induce Retinal and Cardiac Pathology in Drosophila
Read this article at
Abstract
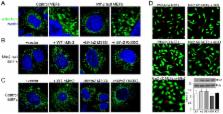
Mitochondrial fusion is essential to organelle homeostasis and organ health. Inexplicably, loss of function mutations of mitofusin 2 (Mfn2) specifically affect neurological tissue, causing Charcot Marie Tooth syndrome (CMT) and atypical optic atrophy. As CMT-linked Mfn2 mutations are predominantly within the GTPase domain, we postulated that Mfn2 mutations in other functional domains might affect non-neurological tissues. Here, we defined in vitro and in vivo consequences of rare human mutations in the poorly characterized Mfn2 HR1 domain. Human exome sequencing data identified 4 rare non-synonymous Mfn2 HR1 domain mutations, two bioinformatically predicted as damaging. Recombinant expression of these (Mfn2 M393I and R400Q) in Mfn2-null murine embryonic fibroblasts (MEFs) revealed incomplete rescue of characteristic mitochondrial fragmentation, compared to wild-type human Mfn2 (hMfn2); Mfn2 400Q uniquely induced mitochondrial fragmentation in normal MEFs. To compare Mfn2 mutation effects in neurological and non-neurological tissues in vivo, hMfn2 and the two mutants were expressed in Drosophila eyes or heart tubes made deficient in endogenous fly mitofusin (dMfn) through organ-specific RNAi expression. The two mutants induced similar Drosophila eye phenotypes: small eyes and an inability to rescue the eye pathology induced by suppression of dMfn. In contrast, Mfn2 400Q induced more severe cardiomyocyte mitochondrial fragmentation and cardiac phenotypes than Mfn2 393I, including heart tube dilation, depressed fractional shortening, and progressively impaired negative geotaxis. These data reveal a central functional role for Mfn2 HR1 domains, describe organ-specific effects of two Mfn2 HR1 mutations, and strongly support prospective studies of Mfn2 400Q in heritable human heart disease of unknown genetic etiology.
Related collections
Most cited references22
- Record: found
- Abstract: found
- Article: not found
The PINK1/Parkin pathway regulates mitochondrial morphology.
- Record: found
- Abstract: found
- Article: not found
Quality control of mitochondria: protection against neurodegeneration and ageing.
- Record: found
- Abstract: found
- Article: not found