- Record: found
- Abstract: found
- Article: found
Unique autosomal recessive variant of palmoplantar keratoderma associated with hearing loss not caused by known mutations*

Abstract
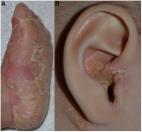
Inherited Palmoplantar Keratodermas are rare disorders of genodermatosis that are conventionally regarded as autosomal dominant in inheritance with extensive clinical and genetic heterogeneity. This is the first report of a unique autosomal recessive Inherited Palmoplantar keratoderma - sensorineural hearing loss syndrome which has not been reported before in 3 siblings of a large consanguineous family. The patients presented unique clinical features that were different from other known Inherited Palmoplantar Keratodermas - hearing loss syndromes. Mutations in GJB2 or GJB6 and the mitochondrial A7445G mutation, known to be the major causes of diverse Inherited Palmoplantar Keratodermas -hearing loss syndromes were not detected by Sanger sequencing. Moreover, the pathogenic mutation could not be identified using whole exome sequencing. Other known Inherited Palmoplantar keratoderma syndromes were excluded based on both clinical criteria and genetic analysis.
Related collections
Most cited references13
- Record: found
- Abstract: found
- Article: found
Olmsted syndrome: clinical, molecular and therapeutic aspects
- Record: found
- Abstract: found
- Article: not found
A molecular defect in loricrin, the major component of the cornified cell envelope, underlies Vohwinkel's syndrome.
- Record: found
- Abstract: found
- Article: not found