- Record: found
- Abstract: found
- Article: found
Quantitative Phylogenomics of Within-Species Mitogenome Variation: Monte Carlo and Non-Parametric Analysis of Phylogeographic Structure among Discrete Transatlantic Breeding Areas of Harp Seals ( Pagophilus groenlandicus)
Read this article at
Abstract
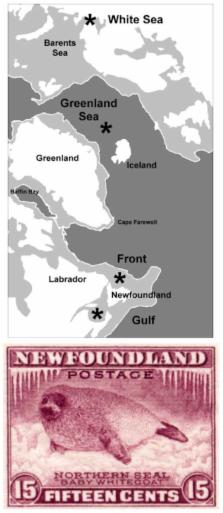
Phylogenomic analysis of highly-resolved intraspecific phylogenies obtained from complete mitochondrial DNA genomes has had great success in clarifying relationships within and among human populations, but has found limited application in other wild species. Analytical challenges include assessment of random versus non-random phylogeographic distributions, and quantification of differences in tree topologies among populations. Harp Seals ( Pagophilus groenlandicus Erxleben, 1777) have a biogeographic distribution based on four discrete trans-Atlantic breeding and whelping populations located on “fast ice” attached to land in the White Sea, Greenland Sea, the Labrador ice Front, and Southern Gulf of St Lawrence. This East to West distribution provides a set of a priori phylogeographic hypotheses. Outstanding biogeographic questions include the degree of genetic distinctiveness among these populations, in particular between the Greenland Sea and White Sea grounds. We obtained complete coding-region DNA sequences (15,825 bp) for 53 seals. Each seal has a unique mtDNA genome sequence, which differ by 6 ~ 107 substitutions. Six major clades / groups are detectable by parsimony, neighbor-joining, and Bayesian methods, all of which are found in breeding populations on either side of the Atlantic. The species coalescent is at 180 KYA; the most recent clade, which accounts for 66% of the diversity, reflects an expansion during the mid-Wisconsinan glaciation 40 ~ 60 KYA. F ST is significant only between the White Sea and Greenland Sea or Ice Front populations. Hierarchal AMOVA of 2-, 3-, or 4-island models identifies small but significant Φ SC among populations within groups, but not among groups. A novel Monte-Carlo simulation indicates that the observed distribution of individuals within breeding populations over the phylogenetic tree requires significantly fewer dispersal events than random expectation, consistent with island or a priori East to West 2- or 3-stepping-stone biogeographic models, but not a simple 1-step trans-Atlantic model. Plots of the cumulative pairwise sequence difference curves among seals in each of the four populations provide continuous proxies for phylogenetic diversification within each. Non-parametric Kolmogorov-Smirnov (K-S) tests of maximum pairwise differences between these curves indicates that the Greenland Sea population has a markedly younger phylogenetic structure than either the White Sea population or the two Northwest Atlantic populations, which are of intermediate age and homogeneous structure. The Monte Carlo and K-S assessments provide sensitive quantitative tests of within-species mitogenomic phylogeography. This is the first study to indicate that the White Sea and Greenland Sea populations have different population genetic histories. The analysis supports the hypothesis that Harp Seals comprises three genetically distinguishable breeding populations, in the White Sea, Greenland Sea, and Northwest Atlantic. Implications for an ice-dependent species during ongoing climate change are discussed.
Related collections
Most cited references16
- Record: found
- Abstract: found
- Article: not found
Mitochondrial genome variation and the origin of modern humans.
- Record: found
- Abstract: found
- Article: not found
Natural selection shaped regional mtDNA variation in humans.
- Record: found
- Abstract: found
- Article: not found