- Record: found
- Abstract: found
- Article: found
Essential site scanning analysis: A new approach for detecting sites that modulate the dispersion of protein global motions
Read this article at
Graphical abstract

Abstract
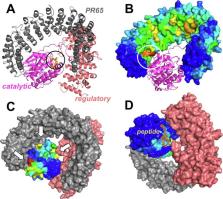
Despite the wealth of methods developed for exploring the molecular basis of allostery in biomolecular systems, there is still a need for structure-based predictive tools that can efficiently detect susceptible sites for triggering allosteric responses. Toward this goal, we introduce here an elastic network model (ENM)-based method, Essential Site Scanning Analysis (ESSA). Essential sites are here defined as residues that would significantly alter the protein’s global dynamics if bound to a ligand. To mimic the crowding induced upon substrate binding, the heavy atoms of each residue are incorporated as additional network nodes into the α-carbon-based ENM, and the resulting shifts in soft mode frequencies are used as a metric for evaluating the essentiality of each residue. Results on a dataset of monomeric proteins indicate the enrichment of allosteric and orthosteric binding sites, as well as global hinge regions among essential residues, highlighting the significant role of these sites in controlling the overall structural dynamics. Further integration of ESSA with information on predicted pockets and their local hydrophobicity density enables successful predictions of allosteric pockets for both ligand-bound and -unbound structures. ESSA can be efficiently applied to large multimeric systems. Three case studies, namely (i) G-protein binding to a GPCR, (ii) heterotrimeric assembly of the Ser/Thr protein phosphatase PP2A, and (iii) allo-targeting of AMPA receptor, demonstrate the utility of ESSA for identifying essential sites and narrowing down target allosteric sites identified by druggability simulations.
Related collections
Most cited references62
- Record: found
- Abstract: found
- Article: not found
Activation and allosteric modulation of a muscarinic acetylcholine receptor
- Record: found
- Abstract: found
- Article: not found
Hydrogen exchange mass spectrometry for studying protein structure and dynamics.
- Record: found
- Abstract: found
- Article: not found