- Record: found
- Abstract: found
- Article: found
Role of autophagy in tumor necrosis factor-α-induced apoptosis of osteoblast cells
Read this article at
Abstract
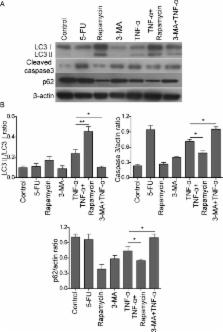
The aim of this study is to investigate the role of tumor necrosis factor-α (TNF-α) in apoptosis and autophagy of mouse osteoblast MC3T3-E1 cells, as well as the crosstalk between autophagy and apoptosis. Mouse osteoblast MC3T3-E1 cells were cultured in vitro and treated with 5-fluorouracil (5-FU), rapamycin, 3-methyl adenine (3-MA) and TNF-α either alone or in combination, respectively. MTT assays were used to monitor the cell viability upon different treatments. Annexin-V-FITC/propidium iodide (PI) staining was used to detect the apoptotic rate of osteoblasts. Autophagic structure and apoptotic bodies were visualized by transmission electron microscopy (TEM). Western blot analysis was performed to detect the autophagic marker LC3-II/I, p62 and apoptotic marker cleaved caspase-3. TNF-α inhibits MC3T3-E1 cell viability in a dose-dependent and time-dependent manner. Annexin-V-FITC/PI staining, coupled with TEM, showed that TNF-α induced cell apoptosis and autophagy in MC3T3-E1 cells. The autophagy inducer rapamycin ameliorated TNF-α-induced apoptosis. In contrast, 3-MA, which is an autophagy inhibitor, caused an exaggerated induction of TNF-α-induced apoptosis. TNF-α upregulated autophagy marker LC3-II/I, but downregulated p62 in osteoblasts. Combined treatment of rapamycin and TNF-α further exaggerated this effect, whereas co-treatment of 3-MA and TNF-α decreased LC3-II/I, but increased p62 compared with TNF-α alone. In addition, TNF-α caused an induction of apoptotic marker cleaved caspase-3. TNF-α-mediated induction of cleaved caspase-3 was downregulated by rapamycin, but upregulated by 3-MA, respectively. TNF-α induced both autophagy and apoptosis in osteoblasts, and upregulated autophagy protects the cell by reducing TNF-α-induced apoptosis.
Related collections
Most cited references16
- Record: found
- Abstract: found
- Article: not found
Autophagy regulates TNFα-mediated joint destruction in experimental arthritis.
- Record: found
- Abstract: found
- Article: not found
Osteoblast programmed cell death (apoptosis): modulation by growth factors and cytokines.
- Record: found
- Abstract: found
- Article: not found