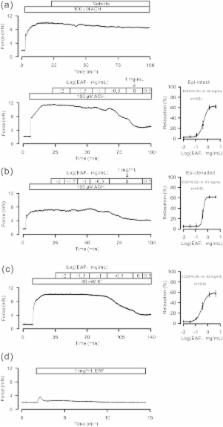
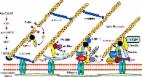
Introduction Airway obstructive diseases (asthma and chronic obstructive pulmonary disease [COPD]) have become increasingly prevalent, currently affecting more than 300 million people worldwide. Dysfunction of airway smooth muscle (ASM) cells, a major cell type in the respiratory tree, plays a pivotal role in promoting progression of these diseases and in contributing to their symptoms of these diseases [1]–[3]. With their ability to contract and relax, these cells regulate the diameter and length of conducting airways, controlling dead space and resistance to airflow to and from gas-exchanging areas. Their excessive contraction, as seen in patients with asthma and COPD, can fully close the airways, thereby preventing gas exchange and threatening life. Not surprisingly, bronchodilators have been used as the medication of choice for asthmatic attacks and as a standard medicine for managing COPD [4],[5]. However, available bronchodilators have adverse side effects, and are not sufficiently effective for severe asthmatics and many other COPD patients. A better understanding of the mechanisms regulating ASM thus holds the promise of developing more effective and safe bronchodilators, which in turn would have a significant impact in reducing mortality and morbidity caused by asthma and COPD. Bitter tastants represent a new class of compounds with potential as potent bronchodilators. Deshpande et al. recently found that cultured ASM cells express G-protein-coupled bitter taste receptors (TAS2Rs) [6], a class of proteins long thought to be expressed only in the specialized epithelial cells in the taste buds of the tongue that allow organisms to avoid harmful toxins and noxious substances characterized by bitterness [7]–[10]. Importantly, bitter tastants with diverse chemical structures cause greater ASM relaxation in vitro than β2 adrenergic agonists, the most commonly used bronchodilators to treat asthma and COPD [6],[11]. Moreover, these compounds can effectively relieve in vivo asthmatic airway obstruction than β2 adrenergic agonists in a mouse model of asthma [6], making them highly attractive bronchodilators for asthma and COPD. Bitter tastant-induced bronchodilation was unexpected, because these agents appeared to increase intracellular Ca2+ concentration ([Ca2+]i) to a level comparable to that produced by potent bronchoconstrictors [6], which should have led to smooth muscle contraction [12]. To reconcile this apparent paradox, it was proposed that bitter tastants activate the canonical bitter taste signaling pathway (i.e., TAS2R-gustducin-phospholipase Cβ [PLCβ]- inositol 1,4,5-triphosphate receptor [IP3R]) to increase focal Ca2+ release from endoplasmic reticulum, which then activate large-conductance Ca2+-activated K+ channels thereby hyperpolarizing the membrane [6]. However, we subsequently demonstrated through patch-clamp recordings that bitter tastants do not activate large-conductance Ca2+-activated K+ channels but rather inhibit them [11]. Moreover, three different large-conductance Ca2+-activated K+ channel blockers did not affect the bronchodilation induced by bitter tastants [11]. Therefore, a different mechanism must be responsible for the bitter tastant-induced bronchodilation. The apparent conundrum of putative [Ca2+]i elevation leading to relaxation may be attributed to the fact that Ca2+ responses to bitter tastants were assessed in cultured human ASM cells, while the contractile responses to them were investigated in freshly dissected ASM tissues [6]. It is well known that cultured smooth muscle cell lines alter their phenotype (i.e., losing their ability to contract and relax [13],[14]) and it is likely their Ca2+ response is also modified. Therefore, to understand bitter tastant-induced bronchodilation, it is necessary to study the contraction and the underlying signaling in freshly isolated ASM tissues and cells. Using this approach in the present study, we found that bitter tastants activate the canonical bitter taste signaling cascade, slightly increasing global [Ca2+]i in resting cells, but not to a level sufficient to cause contraction. However, bitter tastants reverse the increase in [Ca2+]i evoked by bronchoconstrictors, leading to bronchodilation. This reversal is mediated by the suppression of L-type voltage-dependent Ca2+ channels (VDCCs) in a gustducin βγ subunit-dependent, yet PLCβ- and IP3R-independent manner. Hence, we propose that TAS2R activation in ASM stimulates two opposing Ca2+ signaling pathways, both mediated by Gβγ subunits, which increases [Ca2+]i at rest but blocks activated L-type VDCCs reversing the contraction they cause. These results provide the cellular and molecular basis of bitter tastant-induced bronchodilation that occurs in vitro and in vivo. They further reveal a Ca2+ signal that is well suited for screening and identifying potent bronchodilators from among the many thousands of available bitter tastants. Results To uncover the mechanism underlying bitter tastant-induced bronchodilation as demonstrated in both in vitro and in vivo normal and asthmatic models of mice, and in vitro human airways ([6],[11],[15],[16], we examined how bitter tastants affected both [Ca2+]i and ASM contraction in freshly isolated airway cells and tissues from mouse and human. Fluo-3 was used to assess the effect of bitter tastants on [Ca2+]i; chloroquine and denatonium, two substances commonly used to study bitter taste signaling, were used as bitter tastants. Bitter Tastants Modestly Raise Global [Ca2+]i with No Change in Force Generation in Native ASM at Rest We started our analysis by examining the Ca2+ response to bitter tastants in resting cells. In contrast to the marked increase in global [Ca2+]i reported in resting cultured human ASM cells [6], we observed, in resting native ASM cells from mouse, that chloroquine (0.1 µM–1 mM) only modestly raised global [Ca2+]i (and to a level much lower than when cells contracted after application of Mch at 0.1 µM–100 µM) (Figure 1A and Figure S1A). Chloroquine (330 µM) increased fluo-3 fluorescence (ΔF/F0) (i.e., [Ca2+]i) both in the presence of extracellular Ca2+ (37.7%±8%, n = 19) and in its absence (29.3%±6%, n = 15; p>0.05), indicating that the source for this chloroquine response is from internal Ca2+ stores. 10.1371/journal.pbio.1001501.g001 Figure 1 Bitter tastants modestly increase intracellular Ca2+ concentration ([Ca2+]i) by activating a canonical TAS2R signaling cascade. (A) Chloroquine (Chloro) raised [Ca2+]i to a level much less than Mch. [Ca2+]i was measured with fluo-3 in the form of acetoxymethyl ester, loaded into isolated mouse ASM cells, and expressed as ΔF/F0 (%). (B) 1 mM chloro did not contract airways (using tension as its proxy) while 100 µM Mch caused a robust contraction. Data are mean ± SEM (n = 6 for chloro, and n = 5 for Mch). (C) PTX, gallein, anti-βγ (MPS-phosducin-like protein C terminus, a Gβγ blocking peptide), U73122, and 2-APB inhibited chloro-induced increase in [Ca2+]i (n = 19–24 cells). Isolated mouse ASM cells were either pretreated with 1 µg/ml PTX for 6–8 h or with 1 µM anti-βγ for 1–2 h or with each of the other compounds listed for 5–10 min. The effects of PTX and anti-βγ were calculated by normalizing the response of chloro to that from the time matched cells without the pretreatments, and the effects of other three compounds were analyzed by normalizing the response of chloro to its own control without the compound. (D) RT-PCR transcripts after amplification with primers to TAS2R107, α-gustducin, Gβ3, Gγ13, PLCβ2, and β-actin. Note that no transcript was detected with TAS2R108 primers. RNAs were isolated from mouse tracheas and mainstem bronchi, and reactions without complementary DNA were used as a negative control. (E) Cellular distribution of TAS2R107 in three focus planes (bottom, middle, and top) of an isolated mouse ASM cell. The TAS2R107 immunostaining intensity after 3D deconvolution (see Methods) was pseudocolored with the color map on the right. This makes positive (but dim) pixels more easily distinguished from background. Eight cells showed a similar subcellular distribution pattern. To examine whether this modest increase in [Ca2+]i is sufficient to trigger contraction, we measured smooth muscle force formation in mouse airways. As shown in (Figure 1B and S1B), chloroquine (10 µM–1 mM) did not cause contraction of mouse airways, although there was a tendency to decrease the basal tone of airways. As a comparison, Mch at concentrations between 0.3 µM and 10 µM induced contraction markedly and in a dose-dependent manner (Figure 1B and S1B). Bitter Tastants Do Not Generate Localized Ca2+ Events Mouse ASM cells exhibit spontaneous Ca2+ sparks resulting from the opening of ryanodine receptors in the sarcoplasmic reticulum [17]. To test whether bitter tastants generate local Ca2+ events as proposed by Deshpande et al. [6], we stimulated ASM cells with chloroquine (10 µM, a concentration around EC50) for 2 min and measured Ca2+ sparks. Off 40 chloroquine-stimulated cells, 27 cells generated a global [Ca2+]i increase that precluded an accurate estimate of Ca2+ sparks. In the remaining 13 cells without a detectable global rise in [Ca2+]i, chloroquine inhibited the spark frequency but had no effect on the amplitude (frequency [Hz]: 2.13±0.24 in control and 1.62±0.21 with chloroquine [n = 13, p 0.05, paired Student's t-test]). To test whether spontaneous Ca2+ sparks mask the effect of bitter tastants on other forms of local Ca2+ releases, such as Ca2+ puffs due to the opening of IP3Rs [18], we examined the Ca2+ responses to chloroquine in ASM cells pretreated with 100 µM ryanodine. In these cells, prior to chloroquine application, no spontaneous sparks were observed (n = 14). Chloroquine (10 µM) increased global [Ca2+] by 12%±4% (ΔF/F0 at its brightest location) in nine cells, and failed to cause any detectable Ca2+ increase in five cells. There were no detectable local Ca2+ events produced in any of the 14 cells. These results indicate that chloroquine at 10 µM does not increase local Ca2+ events (either Ca2+ puffs or Ca2+ sparks). Bitter Tastants Activate the TAS2R Signaling Pathway to Modestly Raise Global [Ca2+]i in Native ASM at Rest We next examined the cause of the modest global [Ca2+]i rise by bitter tastants. Since in taste cells, bitter tastants bind to TAS2R to activate the pertussis toxin (PTX) sensitive G-protein gustducin, which in turn induces a PLCβ2 and IP3 signaling cascade [19],[20], we studied whether bitter tastants activate this TAS2R signaling pathway. In native ASM cells, PTX (1 µg/ml, and 6–8 h pretreatment), reduced the chloroquine-induced increase in global [Ca2+]i to 21.1%±8.6% of the control cells (n = 20; Figure 1C). Also both gallein (20 µM and 30 min pretreatment), a blocker of the Gβγ dimer of PTX sensitive G proteins, and MPS-phosducin-like protein C terminus, a Gβγ blocking peptide (anti-βγ; 1 µM, and 1 h pretreatment) [21],[22] reduced the bitter tastant-mediated increase in [Ca2+]i to 19.9%±8.5% (n = 19; Figure 1C) and 18.4%±4.8% of the controls, respectively. Finally, U73122 (3 µM), a blocker of PLCβ, and 2-aminoethoxydiphenyl borate (2-APB) (50 µM), an IP3R antagonist, suppressed the bitter tastant-induced increases in [Ca2+]i to 18.0%±5.5% (n = 24) and −10.5%±7.3% of controls, respectively (Figure 1C). These results indicate that bitter tastants do activate the TAS2R signaling transduction pathway (i.e., TAS2R-PTX-sensitive G protein-PLCβ-IP3R) to release Ca2+ from internal stores. This conclusion is further supported by the finding that mouse ASM cells express transcripts for TAS2R107, α-gustducin, Gβ3, Gγ13, and PLCβ2 (Figure 1D), and display peripheral localization of TAS2R107 (Figure 1E). Bitter Tastant-Induced Bronchodilation Is Due to Reversal of the Rise in Global [Ca2+]i Caused by Bronchoconstrictors Bitter tastants at µM levels can modestly increase [Ca2+]i in resting cells, but this raises a conundrum as they also can fully relax airways precontracted by bronchoconstrictors [6],[11]. In light of the fact that an increase in [Ca2+]i is the primary signal for contraction in all smooth muscle, we explored how bitter tastants affect [Ca2+]i evoked by bronchoconstrictors. To better quantify these effects, we measured ASM Ca2+ response and cell shortening at the same time. The cells were stimulated with methacholine (Mch), a stable analogue of acetylcholine, which is the major neurotransmitter in parasympathetic nerves. As expected, Mch (100 µM) rapidly increased [Ca2+]i as fluo-3 fluorescence increased by 162%±26% (ΔF/F0), and concurrently caused cell shortening by 49%±8% (n = 21; Figure 2A and 2B). Strikingly, chloroquine (1 mM) almost completely reversed this [Ca2+]i increase (i.e., bringing [Ca2+]i down to a level only 15%±2% higher than pre-stimulation levels, n = 12, p 0.05 for U73122 and 2-APB. Data are shown as mean ± SEM (n = 12–38 cells). (C) A model for TAS2R signaling and bitter tastant-induced bronchodilation. We propose that bitter tastants activate the canonical TAS2R signaling cascade; this modestly increases [Ca2+]i in resting cells but exerts no significant effect on resting tone. On the other hand, activation of TAS2Rs activates gustducin and release Gβγ, which turns off L-type VDCCs that are pre-activated by bronchoconstrictors, leading to bronchodilation. Discussion Our results demonstrate that bitter tastant's reversal of the rise in [Ca2+]i evoked by bronchoconstrictors is required for its bronchodilation effect. They also reveal that bitter tastants can generate different and opposing Ca2+ signals depending upon the cellular environment. When administered alone to ASM cells at rest, bitter tastants activate the canonical TAS2R signaling pathway to modestly raise [Ca2+]i (Figure 5C) without affecting the contraction. Yet when applied in the presence of the bronchoconstrictors Mch and KCl, they inhibit L-type VDCCs, leading to a reversal of both the evoked [Ca2+]i rise and the contraction (Figure 5C). Remarkably, both types of Ca2+ signals require Gβγ, while only the increase in resting [Ca2+]i depends on PLCβ2 activation and IP3 generation. Bitter taste receptors (35 in mouse and 25 in human) belong to seven transmembrane domain G-protein-coupled receptors. Long thought to only be expressed in the epithelium cells of the taste buds of the tongue, recent studies have revealed that these receptors also express in several extraoral tissues including brain, testis, immune cells, gastrointestinal tract, and respiratory system [50]–[57]. In airways, these receptors are found to be expressed in ciliated epithelial cells and nasal solitary chemosensory cells [51],[52]. Deshpande et al. [6] reported that multiple TAS2Rs can be detected in cultured human ASM cell lines. In this study, for the first time, to our knowledge, we found that this class of G-protein-coupled receptors is expressed in native mouse ASM cells. Specifically, we determined that TAS2R107, to which both chloroquine and denatonium are ligands, localizes in the cell periphery, a location well suited for mediating bronchodilation in response to these two bitter tastants. We cannot rule out that other types of TAS2Rs also contribute to the bronchodilation induced by chloroquine and denatonium, since both of them can activate multiple mouse and human TAS2Rs [58]. Nevertheless, these ligands do activate TAS2R signaling transduction, resulting in a bronchodilation effect, because pharmacological blocking of multiple downstream components of bitter taste receptors can prevent chloroquine and denatonium-induced cellular responses (Figures 1C, 5A, and 5B). It is also worth noting that chloroquine, denatonium, and all the bitter tastants examined so far are not endogenous ligands for bitter taste receptors. Hence a major question remains as to whether bitter taste receptors in ASM cells have physiological ligands. Interestingly, a recent study revealed that acyl–homoserine lactones, quorum-sensing molecules for Gram-negative pathogenic bacteria, can activate bitter tastant receptors in nasal solitary chemosensory cells to evoke trigeminally mediated reflex reactions, which may trigger an epithelial inflammatory response before the bacteria reach population densities capable of forming destructive biofilms [52],[53]. It would be of great interest and significance to investigate whether these quorum-sensing molecules can activate bitter taste receptors in ASM to induce bronchodilation. This study revealed two major differences in Ca2+ signaling compared to the study by Deshpande et al. [6]. First, these authors reported that bitter tastant increased [Ca2+]i to a level comparable to bronchoconstrictors. In freshly isolated ASM, we found that bitter tastants only modestly increase [Ca2+]i to a level much lower than that produced by bronchoconstrictors. Second, Deshpande et al. [6] reported that bitter tastants generate local Ca2+ events. However, in freshly isolated ASM, we found that bitter tastants do not increase local Ca2+ releases such as Ca2+ puffs and Ca2+ sparks. A reason for these two discrepancies may be that Deshpande et al.'s studies were conducted in cultured ASM cell lines; compared to freshly isolated ASM, these cells display a different phenotype by altering the expression of receptors, ion channels, and contractile proteins [13],[14]. The aforementioned two differences and another difference in which we found that bitter tastants do not activate large-conductance Ca2+-activated K+ channels strongly argue that bitter tastant-induced bronchodilation is highly unlikely to result from the generation of local Ca2+ events, which in turn activate large-conductance Ca2+-activated K+ channel and hyperpolarize the membrane as proposed [6]. Since bitter tastants relax precontracted airways [6],[11],[15],[16], it is imperative to use a similar stimulating paradigm in order to understand the underlying mechanism of this relaxation. By simultaneously measuring [Ca2+]i and cell shortening, we found that bitter tastant's ability to reverse the increase in [Ca2+]i caused by bronchoconstrictors is the underlying signal producing the bronchodilation. Three lines of evidence support this conclusion. First, in the presence of bronchoconstrictors, bitter tastants lowered [Ca2+]i while at the same time relaxing the precontracted cells, and this response was reversible. Second, clamping intracellular [Ca2+]i to levels produced by the bronchoconstrictors (low µM) prevented bitter tastants from relaxing airways. Third, enhancing and blocking Ca2+ influx via L-type Ca2+ channels can oppositely regulate the relaxation mediated by bitter tastants. These results reinforce the idea that [Ca2+]i is the critical signal governing ASM contractility. The opposing Ca2+ signals mediated by Gβγ upon activation of TAS2Rs revealed in this study are unique. It is expected that gustducin Gβγ activates PLCβ to generate IP3 and release Ca2+ from endo/sarcoplasmic reticulum to raise [Ca2+]i in ASM cells. But, unexpectedly, gustducin Gβγ also suppresses Ca2+ signaling mediated by Mch, which largely activates M3R, a Gq family receptor. In general, Gβγ from the Gi/Go family (to which TAS2Rs belong) tends to potentiate, rather than, inhibit the Ca2+ responses caused by the Gq family [59],[60]. It remains to be determined whether the inhibition of Ca2+ signaling by TAS2R activation is Gβγ isoform specific. Since Gβγ also mediates the ASM contractions induced by activation of M2R and γ-aminobutyric acid-B receptors [61],[62], our present findings suggested that Gβγ reversal of the rise in [Ca2+]i caused by bronchoconstrictors is isoform specific, and is likely via Gβ3γ13 dimers, which are released upon activation of TAS2Rs [63]. Further studies using ASM cells with genetic deletions of these isoforms should facilitate studying this possibility. It is worthy of mention that virtually all of the studies of bitter taste signaling in taste buds [7]–[10] and extraoral tissues [51]–[53] have focused on the responses mediated by bitter tastants alone; the opposing Ca2+ signaling mediated by Gβγ as revealed in the present study likely operates in these systems when they are stimulated by a combination of bitter tastants and other activators. L-type VDCCs in smooth muscle can be modulated by a variety of means including phosphorylation and Ca2+ [64]–[68]. Yet for the first time, to the best of our knowledge, we show that βγ subunits of G-protein gustducin can inhibit these channels in smooth muscle, extending the similar findings for cloned Cav1.2 in heterologous expression cells and Cav1.1 in skeletal muscle fiber [69],[70]. This interpretation is buttressed by the experiments showing that the contraction mediated by KCl-induced activation of presynaptic Ca2+ channels can also be fully blocked by bitter tastants (Figure 4B). What remains unknown is whether Gβγ directly or indirectly inhibits these channels, and the structural basis for this inhibition. Given that Gβγ can directly inhibit K+ channels and N-type Ca2+ channels in several cell types [71]–[75], it is likely that Gβγ acts on L-type VDCCs in a similar manner. Gustducin βγ subunits inhibit L-type VDCCs to cause bronchodilation, highlighting the importance of these channels in mediating bronchoconstriction and their potential as a target for bronchodilators. Indeed, L-type VDCCs are expressed in ASM cells and their activation causes these cells to fully contract (Figures 4, S4, S5, S6) [17],[27],[76],[77]. Also, activation of these channels is a major mechanism underlying bronchoconstrictor-induced contraction of different species including airway and human ([25],[27],[30],[32], but see [24]). Moreover, three classes of organic L-type VDCC blockers (i.e., dihydropyridines, phenylalkylamines, and benzothiazepines) are effective in relieving airway spasm in animal models of asthma and in exercise-induced asthmatic patients [78]–[81]. A long-standing puzzle regarding L-type channels in ASM is that clinical trials in the 1980s suggested that antagonists for this channel were of limited use treating asthma in the population as a whole [81],[82]. A potential reason for this enigma may be to some extent related to the mode of action of these blockers. It is known that these classic organic blockers exert their inactivation of L-type VDCCs in a voltage, stimulation, and frequency dependent manner [34],[36],[37]. Interestingly, allergen sensitized guinea-pig and rabbit ASM cells have a more hyperpolarized membrane potential than normal cells [83]. This implies that L-type Ca2+ channel blockers (for example, dihydropyridines) would bind more weakly with these channels, thus decreasing the efficacy of these agents to inhibit these channels, should ASM cells from asthma patients have a more negative membrane potential. Bitter tastants, by their ability to inhibit L-type Ca2+ channels via activation of gustducin Gβγ, perhaps could circumvent the drawbacks of the currently available L-type Ca2+ channel blockers, and thus be a more effective asthma treatment. This is likely given that bitter tastants induce a stronger bronchodilation in both in vitro and in vivo asthmatic mouse models than do β2 agonists [6],[11], the most commonly used bronchodilators for treating asthma and COPD. Although bitter tastants are promising candidates to be developed as a new class of bronchodilators, and the findings in the present study provide the cellular and molecular rationale for this line of inquiry, we would caution that chloroquine and denatonium examined in this study may not be ideal candidates because of the high concentration (i.e., on the order of 100 µM) needed to fully relax precontracted ASM. This caveat, however, should not dampen enthusiasm for this endeavor as there are many thousands of bitter tastants available from plants and animals, and numerous bitter small molecules synthesized by research laboratories and a variety of companies over the years. In fact, bitter tastants can stimulate bitter taste receptors at concentrations in the nanomolar range: strychnine activates human TAS2R46 with an EC50 of 430 nM and aristolochic acid activates human TAS2R43 with an EC50 of 81 nM [84],[85]. Therefore, it is highly likely that bitter tastants with a highly potent bronchodilating action can be discovered. Searching for these bitter tastants is of clinical significance because the current bronchodilators are insufficient for treating severe asthma and many COPD patients. A critical step in identifying highly potent bitter tastants is developing reliable and highly effective screening methodologies. Simultaneous measurements of cell shortening and the [Ca2+]i signal (i.e., a decrease of elevated [Ca2+]i), as developed in the present study, are robust and quantitative and provide a powerful paradigm for identifying potential bronchodilators from among the many bitter tastants available. Materials and Methods Animal Tissue Handling Experimental protocols for animal research were approved by the Institutional Animal Care and Use Committees at the University of Massachusetts Medical School (protocol A-1473 to RZG). Isolation of Mouse Airway Smooth Muscle Cells C57BL/6 mice from 7 to 12 wk of age were anesthetized with intraperitoneally injected pentobarbitone (50 mg kg−1), and the trachea and mainstem bronchi were quickly removed and placed in a pre-chilled dissociation solution consisting of (in mM): 135 NaCl, 6 KCl, 5 MgCl2, 0.1 CaCl2, 0.2 EDTA, 10 HEPES, and 10 Glucose (pH 7.3). Tracheas and mainstem bronchi were dissected free from the surface of the connective tissue. The airway tissue was incubated in the dissociation medium containing papain 30 unit/ml, 1 mM DTT, and 0.5 mg/ml BSA, at 35°C for 30 min, and then transferred to a dissociation medium containing 3 unit/ml collagenase F and 0.5 mg/ml BSA, and incubated at 35°C for another 15 min to produce isolated ASM cells. Finally, the tissue was agitated with a fire polished wide-bore glass pipette to release the cells. Mouse Airway Smooth Muscle Contraction Bioassay C57BL/6 mice at 7–12 wk of age were sacrificed and the entire respiratory trees were rapidly removed and immersed in Krebs physiologic solution containing (in mM) 118.07 NaCl, 4.69 KCl, 2.52 CaCl2,1.16 MgSO4, 1.01 NaH2PO4, 25 NaHCO3, and 11.10 glucose. Trachea and mainstem bronchi were isolated and cut into rings (4 mm in length). The rings were mounted on a wire myograph chamber (Danish Myo Technology), and a PowerLab recording device (AD Instruments) was used to record isometric tension. The ring preparations with zero tension were immersed in 5 ml of Krebs physiologic solution, bubbled with 95% O2 and 5% CO2 at 37°C. After 10 min equilibration, three stretches (each 2.5 mN) at 5 min intervals were applied to the rings. After these stretches the basal tones of the rings were usually settled at approximately 2 mN. To test the contractile response, each ring was stimulated twice with KCl (60 mM), separated by 20 min, before proceeding to other treatments. The order and treatment time of agonists and antagonists are indicated in the figure captions. In the experiments in Figures 4B, S4B, and S4D, 1 µM tetrotodoxin was added to prevent action potentials of neurons and 1 µM indomethacin to inhibit cyclooxygenase. The force in response to 60 mM KCl in the presence of tetrodotoxin and indomethacin was 95%±7% (n = 6) of that in their absence. Airway Smooth Muscle Permeabilization Mouse bronchi rings (4 mm in length) free of connective tissues were incubated for 5 min in HEPES-Tyrode (H-T) buffer which contained 137.0 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2, 1.8 mM CaCl2, 10 mM HEPES, 5.6 mM glucose (pH 7.4). The rings were then transferred to and incubated in Ca2+-free H-T buffer for 5 min followed by another 5 min in buffer A (30 mM TES, 0.5 mM DTT, 50 mM KCl, 5 mM K2EGTA, 150 mM sucrose [pH 7.4]). To skin ASM, the rings were incubated for 45 min with α-toxin (16,000 units/ml) in buffer A at room temperature. After the permeabilization, the rings were treated with 10 µM ionomycin for 10 min to deplete intracellular Ca2+ stores. Skinned airway rings were mounted on the wire myograph chamber and washed two times with pCa 9 solution (20 mM TES, 4 mM K2EGTA, 5.83 mM MgCl2, 7.56 mM potassium propionate, 3.9 mM Na2ATP, 0.5 mM dithioerythritol, 16.2 mM phosphocreatine, 15 units/ml creatine kinase [pH 6.9]). The viability of the skinned muscle rings was examined by stimulation with pCa 4.5 solution (20 mM TES, 4 mM CaEGTA, 5.66 mM MgCl2, 7.53 mM potassium propionate, 3.9 mM Na2ATP, 0.5 mM dithioerythritol, 16.2 mM phosphocreatine, 15 units/ml creatine kinase [pH 6.9]) followed by the pCa 9.0 solution. The muscles that could generate sustained contraction in response to the pCa 4.5 solution and fully relax when exposed to the pCa 9.0 solution were used for subsequent experiments. To test the effect of bitter tastants, the viable muscle rings were induced to contract by exposure to pCa 5.5 solution followed by the administration of bitter tastants at the concentrations indicated in Figure 3. Measurement of Global [Ca2+]i and Ca2+ Sparks Fluorescence images using fluo-3 as a calcium indicator were obtained using a custom-built wide-field digital imaging system. The camera was interfaced to a custom made inverted microscope, and the cells were imaged using either a 20× Nikon 1.3 NA for global [Ca2+] measurement or a 60× Nikon 1.4 NA oil for Ca2+ spark measurement. The 488 nm line of an Argon Ion laser provided fluorescence excitation, with a shutter to control exposure duration, and emission of the Ca2+ indicator was monitored at wavelengths >500 nm. The images were acquired at the speed of either 1 Hz for global [Ca2+] measurement or 50 Hz for Ca2+ spark measurement. Subsequent image processing and analysis was performed off line using a custom-designed software package, running on a Linux/PC workstation. [Ca2+]i was represented as ΔF/F0×100 with F calculated by integrating fluo-3 over entire cells for global [Ca2+] after background correction with areas free of cells, or just the value at the brightest pixel (i.e., epicenter pixel) for Ca2+ sparks. Patch-Clamp Recording Membrane currents were recorded with an EPC10 HEKA amplifier under perforated whole-cell patch recording configuration. The extracellular solution contained (in mM): NaCl 126, tetraethylammonium Cl 10, BaCl2 2.2, MgCl2 1, Hepes 10, and glucose 5.6 (pH adjusted to 7.4 with NaOH). The pipette solution contained (in mM): CsCl 139, MgCl2 1, Hepes 10, MgATP 3, Na2ATP 0.5 (pH adjusted to 7.3 with KOH); amphotericin B was freshly made and added to the pipette solution at a final concentration of 200 µg/ml. Whole-cell Ba2+ currents were evoked by step depolarization with 300 ms duration every 10 s from a holding potential of −70 mV at a 10 mV increment (Figure 4A) or with protocol as described in the caption of figure caption (Figure S4C). Currents were leak corrected using a P/4 protocol. Measurement of Cell Shortening Myocytes were placed into a recording chamber superfused with the bath solution for patch clamp experiments at room temperature. Cells loaded with Fluo-3 were imaged using a custom-built wide-field digital imaging system and their lengths were determined using custom software to manually trace down the center of the cell [17]. Reverse Transcription-PCR to Detect mRNA The connective tissues in trachea and mainstem bronchi were carefully removed and the ASM were then quickly frozen in dry ice. The total RNA of the ASM was isolated with the TRIzol (Invitrogen) method following the manufacturer's guidelines; and cDNA was synthesized using extracted RNA with an Omniscript Reverse Transcription kit (Qiagen). The specific primers, synthesized by Invitrogen, are listed in Table S1. β-actin was used as a positive control and the absence of DNA as a negative control, and the PCR reactions were carried out in a PCR mastercycler. Immunocytochemistry Mouse ASM cells, isolated as described above and plated onto poly-L-lysine coated coverslips were fixed and permeabilized (0.1 M ethanolamine in PBS plus 0.1% triton X-100 [pH 8]) and then immunolabeled as described previously [86]. Anti-TAS2R107, an affinity purified rabbit polyclonal antibody raised against a peptide mapping within an extracellular domain of mouse TAS2R107, was purchased from Santa Cruz Biotechnology (sc-139175), and purified IgG was used as control. 3D fluorescence imaging was performed on an inverted wide field microscope (Nikon Diaphot 200) with excitation by a 100 W mercury lamp. Images were obtained through a 60× objective and digitally recorded on a cooled, back-thinned CDD camera (Photometrics), with an effective pixel size at the specimen of 83 nm in x-y and a z spacing of 100 nm. This resulted in a 3D stack of approximately 100 image planes for each cell. The fluorescence images were deconvolved with a constrained, iterative approach [87] originally designed for UNIX systems. The algorithm was rewritten using FFTW, a free, fast Fourier transform library and implemented as a multiuser client/server system on computers running the Fedora operating system (Red Hat), either stand-alone or configured in a Beowulf cluster. Each image was dark current and background subtracted, flat-field corrected, and then deconvolved. After deconvolution images were thresholded to eliminate non-specific binding. Voxels that fell below a threshold were considered to be non-specific bindings and were set to zero; all other voxels remained unchanged. This threshold was derived from analysis of control images containing purified IgG. The intensity which eliminated 99% of the voxels in the control images became the threshold intensity. Reagents and Their Application All chemicals, except fluo-3 (Invitrogen Co), gallein (Tocris Bioscience), anti-βγ blocking peptide (AnaSpec), anti-TAS2R107 (Santa Cruz Biotechnology), and purified IgG (Jackson ImmunoResearch Laboratories) were purchased from Sigma-Aldrich Co. For single cell studies, agonists and antagonists were applied locally to cells via a picospritzer at a constant pressure, so that the duration of its action and concentration could be controlled easily. Statistics Unless stated otherwise, data are reported as mean ± standard error of the mean (SEM) and n represents the number of cells or trachea and mainstem bronchi. Statistical analysis of differences was made with Student's paired or unpaired t-test and the significance level was set at p 0.05 for the response in the first pulse of Mch versus that in the second pulse, Student's paired t-test, n = 10. ΔF/F0 for (B) and (C) are the average over the entire cell. (TIFF) Click here for additional data file. Figure S4 Role of L-type Ca2+ channel activation in Mch-induced contraction in mouse airway. (A) Left panel: L-type VDCC blocker diltiazem dose-dependently reversed Mch-induced contraction (using tension as a proxy measure). Right panel: results for n = 6 airway rings. % relaxation was calculated the same as in Figure S2B. (B) Diltiazem inhibited Mch- and KCl-induced contraction in a dose-dependent manner. (i) Once equilibrated, airway rings generated stable responses to 60 mM KCl (i.e., K0, K1, and K2), and to 1 µM Mch (i.e., M0 and M1) over a time span longer than 1 hr. (ii, iii, iv) show representative responses to KCl and Mch in the presence of diltiazem at 1 µM, 10 µM, and 100 µM, respectively. Two pulses of KCl (K1 and K2) were administrated before Mch (M1) to facilitate diltiazem inactivation of L-type Ca2+ channels. (v) Dose-response curve for diltiazem-mediated inhibition of contraction by KCl. Data are mean ± SEM (n = 5); % inhibition = (force by K0 − force by K2)/(force by K0)×100. (vi) Dose-response curve for diltiazem-mediated inhibition of contraction by Mch. Data are mean ± SEM (n = 5); % inhibition = (force by M0 – force by M1)/(force by M0)×100. Forces are measured at their peak, excluding the noise spikes due to the washing out of KCl or Mch. (C) Diltiazem inhibited L-type VDCC currents in a dose-dependent manner. Diltiazem was added to the bath solution cumulatively. Once at equilibrium at each level of diltiazem, cells were stimulated with a train of ten voltage pulses from −70 mV to 0 mV (inset) at 10 s intervals. Left panel displays patch clamp recordings of L-type Ca2+ currents in the control and in the presence of diltiazem at the given concentration (each in response to the tenth voltage pulse), and the right panel depicts the effect of diltiazem on the peak current at different concentrations. Ba2+ was used as the charge carrier. Data are mean ± SEM (n = 6); % inhibition = (peak current of the control − peak current at given diltiazem concentration)/peak current of the control ×100 (measured in the tenth voltage pulse). (D) Effect of nisoldipine on Mch-induced contraction. In light of the voltage-dependence of the inhibition of nisoldipine on L-type VDCCs, airway rings were modestly depolarized with 20 mM KCl (red bars) before Mch stimulation. Left panel displays a representative contractile response to 60 mM KCl and 1 µM Mch before and after 1 µM nisoldipine. Right panel shows the mean values (mean ± SEM) of inhibition by 1 µM nisoldipine of contraction evoked by KCl and Mch, respectively. % inhibition = (maximal force at the control − maximal for with nisoldipine)/maximal force at the control ×100. (TIFF) Click here for additional data file. Figure S5 KCl activates VDCCs in cholinergic nerves and ASM. (A) Atropine dose-dependently inhibited Mch-induced mouse airway contraction. The left panels display representative contractile responses to 3 µM Mch in the absence or the presence of atropine as marked near the traces, and the right panel shows the mean values (mean ± SEM; n = 4–8) of inhibition by atropine of contraction evoked by 3 µM Mch. % inhibition = (maximal force from the control − maximal force with atropine)/maximal force from the control ×100. (B) Atropine inhibited KCl-induced mouse airway contraction. The left panel shows a representative tension recording in response to 60 mM KCl before and after 100 nM atropine, as marked beneath. The right panel shows the summarized results as the ratio of the force generated by the second KCl pulse over first KCl pulse (mean ± SEM; n = 6 for the time matched controls, n = 28 for atropine). (TIFF) Click here for additional data file. Figure S6 KCl activates L-type VDCCs to increase [Ca2+]i and cause contraction in mouse ASM. (A) KCl failed to generate any global [Ca2+]i increase in the absence of extracellular Ca2+ in isolated single ASM cells. (i) A representative [Ca2+]i response to 60 mM KCl in the presence of extracellular Ca2+. (ii, iii, iv) three examples showing that the same concentration of KCl did not increase Ca2+ in the zero Ca2+ medium. This failure was not due to the depletion of intracellular Ca2+ stores because 10 µM Mch still induced Ca2+ release either as a single peak or as oscillations. Eight cells gave rise to similar responses. ΔF/F0 is the average over the entire cell. (B) KCl (60 mM) caused virtually no increase in tension in the absence of extracellular Ca2+. The airways were placed in the Ca2+ free solution for 15 min before the measurement commenced. Left panel shows a pair of representative recordings and right panel the average results. **p<0.01, Student's paired t-test, n = 6 independent experiments. (C) KCl (60 mM)-induced increase in [Ca2+]i was markedly inhibited by prior application of L-type VDCC blocker diltiazem (100 µM). **p<0.01, Student's paired t-test, n = 9 for each conditions. (D) Diltiazem (100 µM) relaxed 60 mM KCl-induced contraction of mouse airways. Data are mean ± SEM (n = 6 independent experiments), and % relaxation definition and analysis are the same as in Figure S2C. (TIFF) Click here for additional data file. Table S1 Primers for Reverse Transcription-PCR. (TIFF) Click here for additional data file. Movie S1 This clip shows 1 mM chloro reversed the 100 µM Mch-induced increase in [Ca2+]i and cell shortening; the first 120 images of this clip were analyzed and plotted in Figure 2A. The images are displayed as fluorescence intensity (rather than ΔF/F0) because the cell changes its shape dramatically in response to stimuli (and changing thickness makes ΔF/F0 measures misleading). (MOV) Click here for additional data file.