- Record: found
- Abstract: found
- Article: found
Estrogen-related receptor γ causes osteoarthritis by upregulating extracellular matrix-degrading enzymes
Read this article at
Abstract
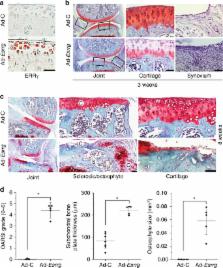
The estrogen-related receptor (ERR) family of orphan nuclear receptor is composed of ERRα, ERRβ, and ERRγ, which are known to regulate various isoform-specific functions under normal and pathophysiological conditions. Here, we investigate the involvement of ERRs in the pathogenesis of osteoarthritis (OA) in mice. Among ERR family members, ERRγ is markedly upregulated in cartilage from human OA patients and various mouse models of OA. Adenovirus-mediated overexpression of ERRγ in mouse knee joint or transgenic expression of ERRγ in cartilage leads to OA. ERRγ overexpression in chondrocytes directly upregulates matrix metalloproteinase (MMP)-3 and MMP13, which are known to play crucial roles in cartilage destruction in OA. In contrast, genetic ablation of Esrrg or shRNA-mediated downregulation of Esrrg in joint tissues abrogates experimental OA in mice. Our results collectively indicate that ERRγ is a novel catabolic regulator of OA pathogenesis.
Abstract
The pathogenesis of osteoarthritis is unclear. The authors show that estrogen-related receptor gamma is upregulated in cartilage from patients and mouse models, where it drives production of matrix-degrading MMPs in chondrocytes, and that its downregulation ameliorates pathology in mice.
Related collections
Most cited references39
- Record: found
- Abstract: found
- Article: not found
Primary culture and phenotyping of murine chondrocytes.
- Record: found
- Abstract: found
- Article: not found
Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction.
- Record: found
- Abstract: found
- Article: not found