- Record: found
- Abstract: found
- Article: found
A Rare Cause of Short Stature: 3M Syndrome in a Patient with Novel Mutation in OBSL1 Gene
Read this article at
Abstract
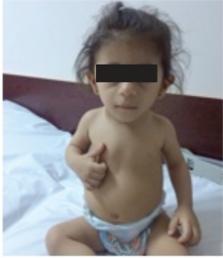
The Miller-McKusick-Malvaux (3M) syndrome is a rare autosomal disorder that can lead to short stature, dysmorphic features, and skeletal abnormalities with normal intelligence. A 16-month-old female patient had been referred to our clinic due to short stature. Case history revealed a birth weight of 1740 grams on the 39 th week of gestation, with a birth length of 42 cm and no prior hereditary conditions of clinical significance in her family. On physical examination, her length was 67 cm [-3.6 standard deviation (SD) score], weight 7.2 kg (-2.9 SD score), and head circumference 42 cm (below 3 rd percentile). She also had numerous characteristic physical features such as a triangular face, fleshy nose tip, a long philtrum, prominent mouth and lips, pointed chin, lumbar lordosis, and prominent heels. As her growth retardation had a prenatal onset and the physical examination results were suggestive of a characteristic profile, the diagnosis of 3M syndrome was strongly considered. Genetic assessment of the patient revealed a novel homozygous p.T45Nfs*40 mutation in the OBSL1 gene. It is recommended that physicians pay further attention to this condition in the differential diagnosis of children with severe short stature.
Related collections
Most cited references6
- Record: found
- Abstract: found
- Article: found
The Genetics of 3-M Syndrome: Unravelling a Potential New Regulatory Growth Pathway

- Record: found
- Abstract: found
- Article: found