1. INTRODUCTION
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease that is typically characterized by persistent synovitis [1] and that manifests as symmetric polyarthritis of large and small joints, which may lead to joint and periarticular structural damage [2]. Although the etiology of RA is unclear, genetic and environmental factors appear to play key roles in the disease [3]. RA affects 0.5–1.0% of the population worldwide [4]; it affects women more often than men, and its prevalence is highest among individuals 35–50 years old [5].
Anti-inflammatory drugs and biological agents are commonly used in RA treatment, but they cannot selectively target inflamed joints, because of their systemic mechanisms, short half-lives and low bioavailability. To circumvent these drawbacks, these agents must be delivered frequently at high doses, thereby increasing the risk of serious adverse effects in healthy joint tissues [6]. Recent research has focused on the development of nanoparticle drug delivery systems that actively or passively target inflamed joints [7] while also improving the release of insoluble drugs, thereby maximizing bioavailability and therapeutic efficacy. Such drug delivery systems can significantly prolong drug half-life in the body and promote drug accumulation in the joints [8]. In this review, we focus on the pathogenesis, pathophysiology and clinical diagnosis of RA, as well as on nanocarriers and other targeted methods that are already used, or that may be used in the future, to treat the disease.
2. THE PATHOGENESIS AND PHYSIOLOGICAL CHARACTERISTICS OF RA
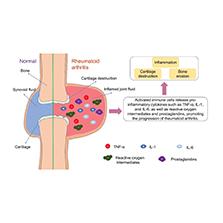
Although the pathogenesis of RA remains unclear, studies have suggested that interactions between genetic and environmental factors lead to innate and adaptive immune responses that promote the development of the disease [9]. External stimuli may also activate innate immune responses by binding Toll-like receptors (TLRs) [10], which activate macrophages, dendritic cells, mast cells, neutrophils, T cells, B cells and fibroblasts [11]. Macrophages drive RA by stimulating neovascularization, clearing apoptotic immune cells, and promoting fibroblast proliferation and protease secretion. Activated macrophages also promote inflammation in RA by releasing pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1 and IL-6, as well as reactive oxygen intermediates and prostaglandins [12]. Strong activation of macrophages also upregulates TLR2, TLR3, TLR4 and TLR7, as well as enzymes, cytokines and other inflammatory factors that promote synovial inflammation and cartilage destruction ( Figure 1 ) [13, 14].

Comparison of normal joints and rheumatoid arthritis joints.
IL, interleukin; TNF, tumor necrosis factor.
Activation of innate immunity causes dendritic cells to promote RA pathogenesis by taking up self-antigens and presenting them to T cells, thus leading to their activation or inhibition [15]. Neutrophils interact with fibroblast-like synoviocytes in the synovium, and consequently promote inflammatory and antigen-presenting phenotypes [16], whereas protein-protein interactions between the receptor activator of nuclear factor (NF)-κB ligand (RANKL) and its receptor, RANK, contribute to osteoclast differentiation in bone remodeling [17]. The RANKL/RANK pathway also promotes inflammation by activating transcription factors and signaling molecules such as NF-κB, JNK, AKT/PKB, ERK, Src kinase and p38 mitogen-activated protein kinase [18]. TNF receptor-associated factors (TRAFs) 1–7 also induce hyperimmune responses that contribute to RA; these factors bind the TNF receptor, IL-1 receptors and TLRs [19]. TRAF6 activates NF-κB, which in turn upregulates expression of genes encoding various inflammatory factors that drive synovitis and the destruction of cartilage and bone [20].
Similarly to tumor tissues, RA tissues are affected by extravasation through leaky vasculature and subsequent inflammatory cell-mediated sequestration (ELVIS), angiogenesis, hypoxia and acidosis. The leaky vasculature of inflamed joints enables the penetration of nanodrugs, which are subsequently internalized by activated synovial cells [21]. The high metabolic demand and rapid growth of synovial membranes in inflamed synovial tissues lead to hypoxia in inflamed joints, thus causing hypoxia-inducible and vascular endothelial growth factors to induce angiogenesis and promote cell growth [22]. Angiogenesis is essential for synovial tissue proliferation and pannus formation. Joint synovial tissues show upregulation of angiogenesis-stimulating factors and downregulation of angiogenesis inhibitors, thus leading to synovial micro-angiogenesis [23]. Gal-9 induces angiogenesis via signaling pathways involving JNK, Erk1/2 and p38 [24]. Survivin, a member of a family of apoptosis inhibitors that block caspase activity [25], upregulates angiogenesis-associated proteins and activates the NOTCH pathway, thereby suppressing apoptosis [26]. Endothelial cells and synovial vessels in inflamed joints contain elevated concentrations of semaphorins, which may contribute to angiogenesis [27]. The pH of interstitial tissues in inflamed joints ranges between 6.0 and 7.0, and acidic pH enhances the inflammatory response of neutrophils [28]. These characteristics of RA can be exploited to achieved targeted treatment of the disease.
3. DIAGNOSIS OF RA
Common diagnostic methods for RA include magnetic resonance imaging (MRI), ultrasonography and assays for autoantibodies. MRI can effectively detect changes in inflamed soft tissues, such as synovitis, tenosynovitis and bone marrow edema, through multiplanar tomographic imaging of bone and soft tissue structures in inflamed joints. MRI can also be used to evaluate cartilage damage, bone erosion and tendon tears of peripheral joints. However, the technique is expensive and time-consuming, and each MRI scan can cover only a limited area of tissue. Ultrasonography, in contrast, enables real-time, relatively low-cost imaging of synovial proliferation and bone erosion in inflamed joints. Ultrasound has similar sensitivity and accuracy to MRI in the diagnosis of synovitis and tenosynovitis in patients with early rheumatoid arthritis [29]. However, ultrasonography cannot detect bone marrow edema. RA can be diagnosed on the basis of the presence of autoantibodies, which are produced in response to abnormal cellular and humoral immune responses. These autoantibodies include rheumatoid factor (RF), which comprises IgG, IgA and IgM, all of which are also present in healthy people but are elevated in individuals with RA; and anti-citrullinated protein antibody (ACPA), which promotes bone loss by activating macrophages or by binding citrullinated vimentin in cell membranes [30]. The concentration of RF and the diversity of its epitopes increase with the levels of pro-inflammatory cytokines. Although RF and ACPA are diagnostic markers of RA, approximately one-third of patients with RA are negative for RF and ACPA. Therefore, simultaneous detection of antibody to RA33, RF and ACPA can improve the diagnostic sensitivity of serological tests [31]. In addition, some researchers have reported that four additional biomarkers—angiotensinogen, serum amylase A-4 protein, vitamin D-binding protein and retinol-binding protein-4—can avoid false-negative findings and improve the accuracy of RA diagnosis [32].
4. DRUGS USED TO TREAT RA
Drugs used to treat RA include non-steroidal anti-inflammatory drugs (NSAIDs), disease-modifying anti-rheumatic drugs (DMARDs), glucocorticoids (GCs), biological agents and RNAs acting through RNA interference (RNAi) ( Table 1 ). These drugs can relieve pain, reduce injury, or efficiently slow the progress of disease [33, 34].
Drugs commonly used to treat rheumatoid arthritis.
| Drug class | Drugs | Adverse effects |
|---|---|---|
| NSAIDs | Acetylsalicylates, naproxen, diclofenac, ibuprofen, etodolac | Nausea, abdominal pain, ulcers, gastrointestinal bleeding, heart failure, high blood pressure |
| DMARDs | Methotrexate, leflunomide, hydroxychloroquine, sulfasalazine, minocycline | Gastrointestinal intolerance (nausea, stomatitis or diarrhea), hepatic toxicity, post-treatment fatigue, headache, dizziness, rheumatoid nodule formation |
| GCs | Dexamethasone, prednisone, prednisolone, betamethasone | Gastrointestinal bleeding, hyperglycemia/diabetes, osteoporosis, infection |
| Biological agents | Infliximab, etanercept, adalimumab, golimumab, certolizumab pegol, tocilizumab, sarilumab | Infection, malignancy, cardiovascular risk, immunogenicity, risk of exposure in pregnancy |
| RNAi | miRNA, siRNA | Rapid enzymatic degradation in the blood, short half-life in serum, low cellular uptake |
DMARDs, disease-modifying anti-rheumatic drugs; GCs, glucocorticoids; miRNA, microRNA; NSAIDs, non-steroidal anti-inflammatory drugs; RNAi, RNA interference; siRNA, small interfering RNA.
NSAIDs are suitable for treatment in early stages of RA; although they can rapidly alleviate symptoms and improve patients’ quality of life, they cannot prevent further joint damage [35]. NSAIDs also inhibit the production of prostaglandins by blocking the ability of cyclooxygenase-2 to transfer arachidonic acid to the endoperoxide pathway, thereby decreasing inflammation associated with RA [36]. Patients’ responses to NSAIDs greatly vary, particularly among older patients. Older patients with RA have an elevated risk of adverse reactions, because they tend to take other drugs in addition to NSAIDs. In addition, older patients tend to have poor compliance with NSAID regimens, as a result of physical dysfunction (such as visual impairment, arthritis, dementia or depression), thus resulting in poor curative effects [37]. Commonly used NSAIDs include acetylsalicylate (aspirin), naproxen, diclofenac, ibuprofen and etodolac. Their long-term use is associated with high risk of nausea, abdominal pain, ulcers, gastrointestinal bleeding, heart failure and high blood pressure [38].
DMARDs, such as methotrexate (MTX), sulfasalazine, hydroxychloroquine, leflunomide and minocycline, can decrease joint swelling, pain and systemic inflammation [35]. MTX is a folic acid analogue that inhibits nucleotide synthesis and purine metabolism by inhibiting the activity of dihydrofolate reductase. This drug was originally used to treat hematological malignancies and has been shown to be effective at low doses in the treatment inflammatory arthritis [34, 39]. Because of its low cost and long-term safety, MTX is currently the most preferred DMARD for RA treatment [40]. However, it has been associated with gastrointestinal intolerance (nausea, stomatitis or diarrhea), hepatotoxicity, post-treatment fatigue, headache, dizziness and rheumatoid nodule formation [41].
GCs, such as dexamethasone, prednisone, prednisolone and betamethasone, are strong anti-inflammatory and immunosuppressive drugs widely used to treat RA [42], because of their ability to control pain, stiffness and swelling. However, GCs are less effective in preventing disease progression [43], and their long-term use increases the risk of cardiovascular disease, gastrointestinal bleeding, hyperglycemia/diabetes, osteoporosis and infection [44, 45].
Biological agents have recently emerged as a novel therapeutic approach for RA, and have been found to effectively alleviate symptoms, slow disease progression and prevent joint injury. Currently, 12 biological agents are used in clinical practice, including five TNF-α inhibitors (infliximab, etanercept, adalimumab, golimumab and certolizumab pegol); inhibitors of IL-6 and its receptor (tocilizumab and sarilumab); a CD80/86-CD inhibitor (abatacept); and an anti-CD20 antibody (rituximab) [34, 46]. However, the use of biological agents has been associated with elevated risk of infection, malignancy, cardiovascular injury, immunogenicity and other adverse events [47].
MicroRNAs (miRNAs) and small interfering RNAs (siRNAs) are small RNA molecules that downregulate protein expression by triggering the degradation of messenger RNAs before their translation into proteins, through a process known as RNAi [48, 49]. In rat models of arthritis, miR-449 has been found to inhibit the production of the inflammatory factor IL-6, whereas miR-708-5p blocks inflammatory cell infiltration, synovial hyperplasia and cartilage destruction [50]. Although siRNA may show promise for therapeutic RNAi, siRNAs are rapidly degraded in the blood and are inefficiently internalized by cells, thus limiting their application in clinical settings [51, 52].
The efficacy of these drugs may be improved through use in combination [53, 54]. Nevertheless, the systemic mechanisms of these drugs and their poor accumulation in inflamed joints require them to be administered often and at high doses, thus increasing the risk of adverse effects [55].
5. TARGETED DRUG DELIVERY SYSTEMS
5.1. Drug delivery systems used to treat RA in animal models
To overcome the disadvantages of conventional RA drugs, several targeting nanodelivery systems are being developed [56, 57] to control drug release and prolong drug circulation in the blood [58] while decreasing systemic toxicity. Nanoparticles, liposomes and micelles are commonly used in medical applications for drug delivery, diagnostics and imaging [59], because of their good biodegradability and sustainability [60] ( Table 2 and Figure 2 ). These materials can stabilize drugs, control drug release and enhance drug accumulation at inflamed sites [61, 85].
Drug-loaded nanoparticle drug delivery vehicles with the potential to treat RA.
| Drug | Carrier type | Brief description | Route of administration | Animal model |
|---|---|---|---|---|
| Dex | Liposomes | Prolonged half-life | Intravenous injection | AIA rats [61] |
| Dex | Liposomes | Enhanced targeting effect | Intravenous injection | AIA rats [62] |
| Tofacitinib citrate | Liposomes | Enhanced distribution in inflamed sites | Intravenous injection | AIA rats [63] |
| Berberine | Liposomes | Diminished inflammatory response | Intravenous injection | AIA rats [64] |
| MTX | Gold NPs | Improved efficacy and diminished toxicity | Intravenous injection | AIA rats [65] |
| Indomethacin | Micelles | Enhanced anti-inflammatory activity | Intravenous injection | AIA rats [66] |
| Dex/palmitate | Micelles | Enhanced targeting effect | Intravenous injection | AIA rats [67] |
| Tacrolimus | Micelles | Enhanced targeting effect | Intravenous injection | AIA rats [68] |
| microRNA124/MTX | Micelles | Enhanced synergy | Intravenous injection | AIA ats [69] |
| Dex/p65 siRNA | Micelles | Co-delivery of siRNA and Dex into macrophages | Intravenous injection | CIA mice [70] |
| Dex/palmitate | PLGA NPs | Improved pharmacokinetics and decreased cell damage | Intravenous injection | CIA mice [71] |
| siRNA | PLGA NPs | Retarded progression of inflammation | Intravenous injection | CIA mice [72] |
| β-Sitosterol | Solid lipid NPs | Inhibition of the NF-κB signaling pathway | Intravenous injection | AIA rats [73] |
| Prednisolone | Solid lipid NPs | Specific binding to CD44 on inflammatory cells | Intravenous injection | CIA mice [74] |
| Embelin | Chitosan NPs | Prolonged retention time and improved targeting | Intravenous injection | AIA rats [75] |
| Eugenol | Chitosan NPs | Enhanced anti-arthritic activity | Intravenous injection | CIA mice [76] |
| Zinc gluconate | Chitosan NPs | Enhanced targeting effects | Intravenous injection | CIA mice [77] |
| Carvacrol | Albumin NPs | Potent suppressive effects | Intravenous injection | AIA rats [78] |
| Celastrol | Albumin NPs | Decreased celastrol dose with effective therapy | Intravenous injection | AIA rats [79] |
| Methotrexate | Albumin NPs | Enhanced retention and decreased carvacrol dose | Intravenous injection | CIA mice [80] |
| Prednisolone/curcumin | Albumin NPs | Co-delivery of prednisolone and curcumin to inflamed sites | Intravenous injection | AIA rats [81] |
| Hydroxychloroquine | Biomimetic NPs | Increased half-life and targeting to inflamed joints | Intravenous injection | CIA mice [82] |
| Dex | Injectable hydrogel | Enhanced therapeutic efficacy | Intra-articular injection | CIA mice [83] |
| Indomethacin/MTX/MMP-9 siRNA | Injectable hydrogel | Synergistic treatment with multiple drugs | Intra-articular injection | AIA rats [84] |
AIA, adjuvant-induced arthritis; CIA, collagen-induced arthritis; NPs, nanoparticles; Dex, dexamethasone; MTX, methotrexate; PLGA, poly (lactic-co-glycolic acid).

Nanoparticles commonly used in targeted delivery systems.
PLGA, poly(lactic-co-glycolic acid).
5.1.1 Liposomes
Liposomes consist of phospholipids and cholesterol, which form a lipid bilayer with an aqueous core. Their particle sizes are usually in the range of 25 nm to 2.5 μm [86]. Liposomes can encapsulate both hydrophobic and hydrophilic drugs, and they have good biocompatibility and biodegradability. However, the ability of liposomes to encapsulate hydrophobic drugs is not ideal, and drugs can easily leak out [87]. Although traditional liposomes are rapidly cleared by the reticuloendothelial system, modifying liposomes with polyethylene glycol (PEG) effectively decreases the adsorption of plasma proteins and subsequent clearance by the reticuloendothelial system, thus prolonging the circulation of drugs in the blood and improving their distribution in inflamed joints [88]. Intravenous administration of dexamethasone-loaded polymerized stealth liposomes to arthritic rats has been found to significantly prolong drug circulation in the blood and to enhance drug accumulation in inflamed joints [61]. This treatment significantly decreases the levels of TNF-α and IL-1β at lesion sites as well as the degree of joint swelling, thus indicating inhibition of RA progression.
The peptide ART-2 (CKPFDRALC) shows preferential homing to arthritic joints of rats and strong binding to endothelial cells. To improve the targeting efficiency of liposomes to inflamed joints, one study has designed liposomes modified with ART-2 peptide. These dexamethasone-loaded liposomes with ART-2 modification accumulate in inflamed joints to a greater extent than dexamethasone-loaded liposomes without ART-2 modification, and relieve RA more efficiently [62]. Beyond hydrophobic drugs, liposomes can also be used to efficiently encapsulate hydrophilic drugs. Hydrophilic drugs can be encapsulated within the aqueous core of liposomes. Liposomes loaded with tofacitinib citrate, a water-soluble anti-inflammatory drug, have been found to be selectively internalized by inflammatory cells in a rat model of arthritis and to accumulate in arthritic paws [63]. This method of tofacitinib citrate delivery significantly improves its therapeutic efficacy, downregulates inflammatory cytokines in joint tissues and relieves RA symptoms. In another study, PEGylated liposomes loaded with water-soluble berberine have been found to accumulate selectively in inflamed joints of rats with adjuvant-induced arthritis (AIA). Berberine potently activates miR-23a, thus downregulating inflammatory kinases such as ASK1 and GSK-3β, as well as mediators of Wnt1 signaling, and ultimately mitigating bone erosion [64]. The safety and efficacy of liposomal bupivacaine has been confirmed in surgery; however, its pharmacokinetic parameters and safety in a Chinese population have not been evaluated. A phase I study has confirmed that liposomal bupivacain is well tolerated and safe among individuals of Chinese descent [89].
5.1.2 Gold nanoparticles
Gold nanoparticles (AuNPs), with particle sizes ranging from 1 to 100 nm, are widely used in diagnostics, therapy and biological imaging [90]. AuNPs have excellent stability and biocompatibility, customizable shapes and dimensions, easily functionalized surfaces, high drug loading capacity and low toxicity. However, AuNPs tend to accumulate in the kidneys, liver and spleen after entering the body, thus potentially leading to incomplete metabolism in the body [91]. AuNPs have strong affinity to thiol and amine groups, and therefore can bind targeting agents possessing these groups. In addition, AuNPs have good binding ability toward vascular endothelial growth factor and show natural antiangiogenic effects in inflamed synovium. Intra-articular injection of AuNPs of various dimensions into CIA mice has indicated clear antioxidant action, by significantly increasing catalase activity without causing any adverse effects on hematological indices. AuNPs of 50 nm, compared with 13 nm, have shown superior effects in inhibiting synovial angiogenesis, and have achieved better antioxidant and therapeutic effects in early stages of arthritis [92]. In another study, an MTX delivery system consisting of gold nanorods with a mesoporous silica shell (FAGMs) has been used for controlled release of MTX. The release rate of MTX from FAGMs in vitro markedly increases under 808 nm laser irradiation, thus achieving superior effects in inhibiting RA progression in AIA rats while decreasing the systemic toxicity of MTX, compared with free MTX [65]. These findings suggest that FAGMs may hold promise in the treatment of RA. In addition, clinical trials have tested the coupling of human proinsulin peptide (C19-A3) with AuNPs (C19-A3-AuNPs) for the treatment of type 1 diabetes. In a phase I clinical trial, the safety of intradermal administration of C19-A3-AuNP through microneedles has been explored [93]. Patients with type 1 diabetes have shown good tolerance to C19-A3-AuNPs with no signs of systemic allergy.
5.1.3 Polymeric micelles
Polymeric micelles are core-shell structures with a particle size of 10–100 nm that form through self-assembly of amphiphilic block copolymers in aqueous solutions [94]. Polymeric micelles can effectively avoid clearance by the kidneys and reticuloendothelial system, and they can target inflamed tissues through the ELVIS effect [95]. Micelles form when the polymer concentration in the solution exceeds the critical micellar concentration, and they dissociate into monomers when the polymer concentration is below the critical micellar concentration. Micelles with lower critical micellar concentrations are therefore more stable in circulation [96].
Copolymeric micelles loaded with indomethacin, a non-steroidal anti-inflammatory drug that can effectively control inflammation [66], have been found to significantly relieve inflammatory symptoms and decrease the arthritic index and paw diameter in AIA rats. Similarly, micelles loaded with dexamethasone palmitate have been found to accumulate at inflamed sites and to decrease joint inflammation [67]. In another study, maltodextrin-α-tocopherol nano-micelles loaded with tacrolimus (TAC@MD-α-TOC) have been prepared for RA therapy. The micelles have been found to show stronger anti-rheumatic effects than the free drug. In vitro and in vivo experiments have demonstrated that TAC@MD-α-TOC is more effective than the free drug in promoting the viability of Vero cells and decreasing the levels of IL-6 and TNF-α in the serum and synovial fluid [68]. Monotherapy with MTX usually leads to irreversible joint injury because of its slow onset and long duration. MicroRNA-124 (miR-124) has shown direct bone protective effects against RA. Hybrid micelles containing both MTX conjugated polymer and miR-124 have been found to target and accumulate in the inflamed joints of AIA rats, and effectively enhance the synergistic effects of MTX and miR-124 [69]. Wang et al. have used polymeric micelles to co-deliver Dex and siRNA against p65, a member of the NF-κB family, to arthritis sites to treat RA [70]. These co-loaded micelles have been found to efficiently inhibit NF-κB signaling in macrophages in CIA mice and to cause activated macrophages in arthritic synovial membranes to revert to an anti-inflammatory state. In the treatment of RA, micelles co-loaded with Dex and p65 siRNA have shown better efficiency than free Dex or naked siRNA. In addition, a phase II study on the efficacy and safety of docetaxel-PM for treating recurrent or metastatic squamous cell carcinoma of the head and neck was conducted in 2015, and this docetaxel-PM is now on the market.
5.1.4 PLGA nanoparticles
Poly(lactic-co-glycolic acid) (PLGA) is a non-toxic, biodegradable polymer often used as a drug delivery carrier. PLGA nanoparticles can regulate drug release, and their surfaces can be easily modified. They are commonly used to protect biological agents, such as proteins and nucleic acids, against rapid metabolism and clearance in vivo [97]. The drug release from PLGA particles depends on the molecular weight of the polymer. Polymers with higher molecular weights have longer polymer chains, thus resulting in longer degradation times and slower drug release rates [98]. For example, loading of apremilast, a small-molecule drug designed for oral delivery, into PLGA nanoparticles significantly prolongs its half-life and mean residence time in vivo [99]. Luteinium-177, a radionuclide with a half-life of 6.71 d, can be used for radiation treatment of joints with advanced arthritis, owing to its β maximum emission energy (0.497 MeV, 78%). Hyaluronic acid (HA) is a polymer that specifically binds CD44 overexpressed on inflamed synovial cells. In one study, PLGA modified with Luteinium-177 and HA has been used for encapsulating MTX (177Lu-DOTA-HA-PLGA(MTX)). The 177Lu-DOTA-HA-PLGA(MTX) nanoparticles strongly bind, and are efficiently internalized by, inflamed synovial cells [100]. In addition, encapsulating dexamethasone palmitate in PLGA-PEG nanoparticles has been found to improve the drug’s pharmacokinetic profile, and decrease its tendency to damage cells and aggregate in the liver, kidneys and lungs [71]. Bruton’s tyrosine kinase (BTK) in macrophages and B cells is an important target in RA therapy. However, high dosages of BTK inhibitors are needed for effective BTK inhibition, thus limiting their clinical application. Zhao et al. have developed cationic lipid-assisted PEG-b-PLGA nanoparticles (CLAN) loaded with BTK siRNA (CLANsiBTK) [72]. In the CIA mouse model, CLANsiBTK has been found to significantly alleviate arthritis symptoms, downregulate the expression of inflammatory cytokines (TNF-α, IL-1β and IFN-γ) and decrease damage to the paw joints.
5.1.5 Solid lipid nanoparticles (SLNs)
SLNs, a lipid-based drug delivery system with a particle size of 10–1000 nm, specifically target inflamed tissues and enable controlled drug release [101]. SLNs show low immunogenicity in the human body and can easily infiltrate biological tissues while carrying large amounts of lipophilic compounds. However, the ability of SLNs to encapsulate hydrophilic drugs is poor. Moreover, changes in the drug release curve, polymorphic transformation and particle aggregation occur during storage [102]. Zhang et al. have loaded β-sitosterol into SLNs to improve its water solubility and bioavailability in AIA rats. The developed nanoparticles (β-sitosterol-SLNs) have been found to exhibit good anti-arthritic effects by inhibiting NF-κB and activating the heme oxygenase-1/NF-erythroid 2-associated factor 2 pathway [73]. To improve the targeting of inflamed tissue, prednisolone-loaded SLNs coated with HA have been prepared; these SLNs have been found to accumulate in the inflamed joints of CIA mice and to decrease joint swelling, bone erosion and levels of inflammatory cytokines in the serum [74].
5.1.6 Chitosan nanoparticles
Chitosan, a polysaccharide that arises through the deacetylation of chitin, is widely used in the preparation of microparticles and nanoparticles [103]. Chitosan nanoparticles have good biodegradability, low immunogenicity and high cell permeability, and therefore are suitable nanocarriers for targeted drug delivery [104]. For example, chitosan nanoparticles can improve the efficacy of the anti-inflammatory drug embelin, which is poorly absorbed in the body, and is rapidly metabolized and cleared. Loading embelin into chitosan nanoparticles downregulates malondialdehyde and nitroxide, as well as TNF-α, IL-6 and IL-1β in the serum in AIA rats, while decreasing oxidative stress [75]. In a study in CIA rats, eugenol loading into chitosan nanoparticles has been found to significantly improve the drug’s ability to decrease the expression of monocyte chemoattractant protein-1 and transforming growth factor-β, and to alleviate joint synovial hyperplasia and cartilage injury [76]. Similarly, loading zinc gluconate into chitosan nanoparticles has been found to improve the compound’s ability to inhibit the infiltration of inflammatory cells in ankle joints; downregulate TNF-α, IL-6 and inducible nitric oxide synthase; and upregulate SOD1 [77].
5.1.7 Albumin nanoparticles
Albumin is the most abundant protein in the plasma, accounting for approximately 60% of the total protein in the blood [105]. It is considered an ideal candidate for drug delivery because of its strong ability to bind hydrophobic and hydrophilic drugs, relatively long half-life in the blood (19 days), biodegradability and lack of immunogenicity [106]. Albumin, which has a molecular weight of 66.5 kDa, can be obtained from various sources, such as egg white (ovalbumin), bovine serum (BSA) or human serum (HSA) [107]. The nanoparticles can prolong the circulation in the blood by adsorbing albumin [108]. For example, coating albumin on the surfaces of liposomes or embedding it directly in phospholipids of liposomes enables the nanostructures to evade phagocytosis, thus prolonging their circulation [109]. In a study in AIA rats, loading carvacrol into BSA nanoparticles has been found to significantly improve the anti-inflammatory agent’s ability to mitigate swelling and decrease release of the inflammatory cytokines NO and IL-17 in arthritic rats [78]. To improve the targeting of BSA nanoparticles, Gong et al. have prepared palmitic acid modified BSA nanoparticles (PAB NPs) and loaded them with celastrol. The PAB NPs efficiently bind scavenger receptor-A (SR-A) and elicit 9–10 times more macrophages than normal BSA NPs. PAB NPs have been found to deliver the anti-inflammatory drug celastrol to inflamed tissues more effectively than BSA NPs, and to alleviate RA symptoms at lower doses [79]. Loading MTX into HSA nanoparticles labeled with chlorin e6 has been found to increase the drug’s accumulation and retention in inflamed joints of CIA rats; the encapsulated drug slows RA progression as effectively as a 50% higher dose of free drug [80]. In a study in AIA rats, treatment with prednisolone and curcumin loaded into HSA nanoparticles has led to lower levels of pro-inflammatory cytokines in activated macrophages, higher levels of the anti-inflammatory cytokine IL-10, greater drug accumulation in inflamed joints and stronger therapeutic efficacy than nanoparticles loaded with single drugs or free drugs alone or a mixture of two drugs [81]. Abraxane (paclitaxel combined with albumin) was approved by the FDA in 2005. In 2021, the FDA approved Fyarro (sirolimus albumin-bound nanoparticles, nab-sirolimus, ABI-009) to be marketed for intravenous infusion in the treatment of locally advanced unresectable or metastatic malignant perivascular epithelioid cell tumors.
All the above studies used exogenous albumin to prepare nanoparticles in vitro. However, after nanoparticles enter the body, they can cause immunogenic reactions. Therefore, some researchers have attempted to use the endogenous albumin in the body to prepare albumin-bound nanoparticles for targeting therapy. The albumin-binding domain (ABD, 46 amino acids) has been reported to have strong affinity for serum albumin. The ABD035 variant has shown superior affinity toward albumin from various sources (such as human, mouse, rat, cow and monkey albumin). Zhang et al. have designed a redox-responsive paclitaxel micelle system modified by the ABD035 peptide [110]. After intravenous injection, the micellar system has been found to combine with endogenous albumin in blood, then deliver paclitaxel to tumor cells via gp60 and SPARC receptors. Inflammatory tissue has similar characteristics to tumor tissue, such as abundant albumin receptors and incomplete vascular structure. Therefore, developing nanoparticles that bind endogenous albumin may serve as a favorable strategy for targeting RA.
5.1.8 Biomimetic nanoparticles
Biomimetic nanoparticles have attracted particular attention in recent years because of their ability to evade clearance by the reticuloendothelial system [111]. Biomembranes are usually extracted through the following method. Cells are placed in hypotonic liquid for cell disruption and lysis, then purified and collected by discontinuous gradient centrifugation at 4 °C. Protease inhibitor is added throughout the entire extraction process to protect protein activity [112]. Nanoplatforms are prepared by encapsulating nanoparticles into cell membranes extracted from red blood cells, macrophages, neutrophils or platelets [113] through co-extrusion, extrusion/sonication, freeze-thaw/sonication, extrusion/sonication and other methods [114, 115]. These nanoplatforms have shown excellent biocompatibility, effective drug delivery, prolonged circulation times in the blood and minimal adverse immune responses [116, 117]. For example, spherical, prolate-spheroidal and oblate-spheroidal PLGA nanoparticles coated with erythrocyte membrane effectively evade clearance by macrophages [118]. Encapsulation of hydroxychloroquine-loaded nanoparticles in membranes from umbilical vein endothelial cells expressing TNF-associated apoptosis-inducing ligand has generated nanoparticles for the delivery of hydroxychloroquine to inflamed joints in CIA mice while also inducing M1 macrophage apoptosis by upregulating death receptor-5 [82], thus effectively inhibiting RA progression. In CIA mice, coating nanoparticles with neutrophil membranes and then administering these nanostructures together with immunoregulatory molecules has been found to promote tissue repair, downregulate pro-inflammatory cytokines and inhibit synovitis [119].
5.1.9 Injectable hydrogels
Hydrogels are highly hydrated three-dimensional networks of hydrophilic polymers, whose bionic structure is similar to that of the extracellular matrix of natural biological tissues and has good biocompatibility. Injectable hydrogels are more comfortable, and elicit less pain and fewer adverse effects, than non-injectable hydrogels. Favorable injectable hydrogels cannot easily be obtained by using a single material. Incorporating nanofiller into a polymer matrix can achieve desirable injectable gels with high biocompatibility and biodegradability, more easily modifiable properties, and better ability to deliver hydrophilic or hydrophobic macromolecules in a sustained manner. Injectable hydrogels with loosely interconnected polymer chains have shown higher burst release and more rapid drug clearance [120]. Wang et al. have developed a temperature-sensitive hydrogel (DLTH) based on chitosan-glycerol-borax for intra-articular delivery of dexamethasone [83]. In the CIA mouse model, intra-articular injection of DLTH loaded with dexamethasone has shown good anti-inflammatory and analgesic effects. Similarly, in situ hydrogels have also been designed to co-deliver indomethacin, methotrexate and MMP-9 siRNA for synergistic and comprehensive treatment of RA [84]. These in situ hydrogels significantly down-regulate the expression of inflammatory factors (TNF-α, IL-6) and MMP-9 in plasma and knee joint fluid after intra-articular injection.
5.2. Targeted RA treatment in animal models
5.2.1 Passive targeting
Synovial thickening during the development of RA induces hypoxia and angiogenesis, thus leading to vascular leakage at sites of inflammation. Because the ELVIS effect in RA is similar to the EPR effect in tumor tissues [21], as exploited by many cancer therapies, passive targeting strategies against RA have been developed on the basis of the ELVIS effect [121]. Nanoparticles of 20–200 nm can penetrate the synovial tissue through the intercellular space between endothelial cells and accumulate at inflamed sites, thus significantly limiting drug adverse effects ( Figure 3 ). In contrast, nanoparticles smaller than 10 nm are easily cleared by renal filtration, whereas those larger than 200 nm are removed by phagocytes in the reticuloendothelial system [122, 123].

Schematic illustration of the extravasation through leaky vasculature and subsequent inflammatory cell-mediated sequestration (ELVIS) effect.
To improve the passive targeting ability of nanoparticles of suitable size, their surfaces have been modified with PEG, which shields surface charge, inhibits the adsorption of serum proteins and helps evade detection by circulating macrophages [124, 125]. For example, allowing hydrophilic short-chain methoxy PEG to self-assemble with gambogic acid, a natural drug that inhibits inflammation by downregulating IL-1β and TNF-α, has been found to increase the drug’s ability to ameliorate paw inflammation in CIA mice [126]. The free drug, in contrast, shows low water solubility, poor pharmacokinetics and hemolytic toxicity. Similarly, modifying liposomes 100 nm in diameter with slightly negative charge with 10% PEG5000 has been found to increase their in vivo circulation time and ability to target inflamed joints in CIA mice, thus resulting in much better effects of encapsulated dexamethasone than the free drug [127]. Similarly, liposomes prepared with hydrogenated phosphatidylinositol can avoid clearance by the reticuloendothelial system and prolong the blood circulation time. Doxorubicin loaded in hydrogenated phosphatidylinositol liposomes has been detected within 72 hours after injection. The circulation time of hydrogenated phosphatidylinositol liposomes is significantly longer than that of ordinary liposomes [128]. In addition, nanoparticles coated with poly(ethylene oxide)-block-poly (γ-methylprednisolone) have nearly neutral surface charges, can avoid the non-specific binding of macromolecules to nanoparticles, and additionally can escape clearance by reticuloendothelial systems [129].
5.2.2. Active targeting
Active targeting primarily involves modifying the nanocarrier surface with appropriate ligands that bind receptors expressed on the surfaces of target cells at inflamed sites ( Table 3 ) [140]. RA involves vascular regeneration and inflammation, and these processes involve B cells, T cells, macrophages and other immune cells [11]. These processes are driven by growth factors, pro-inflammatory cytokines, chemokines, cell adhesion molecules, proteases and the hypoxia-vascular endothelial growth factor angiopoietin. Activated macrophages are abundant at sites of inflammation, where they produce large amounts of pro-inflammatory IL-6, IL-1β and TNF-α [141]. The surfaces of activated macrophages contain abundant folate receptor-β, vasoactive intestinal peptide receptor, scavenger receptor class A, TLRs, transforming growth factor-β receptor, CD44, CD64 and other receptors ( Figure 4 ), whereas the surfaces of endothelial cells contain abundant αvβ3-integrin, E-selectin, vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 [142, 143]. If drug-loaded nanoparticles can selectively bind these receptors, they can target their cargo to inflamed tissue, provided that the nanoparticles can evade clearance by the reticuloendothelial system and penetrate the inflamed vascular endothelium ( Figure 5 ) [55].
Drug-loaded nanoparticles targeting receptors highly expressed in rheumatoid arthritis tissue.
| Targeted receptor | Drug | Carrier | Animal model |
|---|---|---|---|
| Folate receptor | MTX | Chitosan nanoparticles | CIA mice [130] |
| Folate receptor | MTX | Liposomes | CIA mice [131] |
| Folate receptor | Myeloid cell leukemia-1 siRNA/Dex | Micelles | CIA mice [132] |
| Scavenger receptor | MTX | Double layered hydroxide nanocomposites | AIA rats [133] |
| Scavenger receptor | Celastrol | Micelles | AIA rats [134] |
| CD44 | Dex | Polymeric nanoparticles | AIA rats [135] |
| CD44 | MTX/teriflunomide | Hydroxyapatite nanoparticles | CIA mice [136] |
| CD44 | Tripterine | Bilosomes | CAIA rats [137] |
| Mannose receptor | p-Coumaric acid | Liposomes | AIA rats [138] |
| Mannose receptor | Morin | Liposomes | AIA rats [139] |
AIA, adjuvant-induced arthritis; CAIA, collagen antibody-induced arthritis; CIA, collagen-induced arthritis; MTX, methotrexate; Dex, dexamethasone.

Active targeting of cells in inflamed joints through specific binding between ligand-modified nanoparticles and targeted receptors.

Active targeting of inflamed tissues by (1) drug-loaded nanoparticles modified with specific ligands that can evade (2) reticuloendothelial clearance and penetrate (3) inflamed vascular endothelium.
Folate receptors are glycopolypeptides with high affinity for folic acid that have four isoforms (FR-α, FR-β, FR-γ and FR-δ) that are differentially expressed among tissues [144]. FR-β is widely expressed on the surfaces of activated synovial macrophages in patients with RA and therefore may be a potential target for disease treatment [145]. MTX encapsulated in hydrophobically modified ethylene glycol chitosan nanoparticles conjugated to folic acid, compared with nanoparticles without folic acid conjugation, has been found to result in significantly lower pro-inflammatory cytokine expression, paw thickness and arthritic scores in CIA mice [130]. In another study, folic acid has been coupled with PEG100 monostearate to prepare liposomes co-loaded with MTX and catalase. The obtained liposomes show enhanced cellular uptake through folate-mediated endocytosis and strong toxicity against activated RAW264.7 cells, as well as enhanced accumulation in inflamed joints and therapeutic efficacy in CIA mice, while causing minimal toxicity to major organs [131]. Myeloid cell leukemia-1 (Mcl-1) is overexpressed in the macrophages of arthritic joints. Inhibiting MCL-1 induces apoptosis of macrophages and thus alleviates inflammation. Li et al. have developed folate-modified polymeric micelles co-loaded with MCL-1 siRNA and DEX for RA therapy [132]. In the CIA mouse model, these DEX/siRNA co-loaded polymeric micelles have been found to significantly decrease MCL-1 mRNA levels in macrophages, as well as levels of inflammatory factors such as TNF-α and IL-1β.
Scavenger receptors belong to a superfamily of structurally heterogeneous proteins, including transmembrane proteins and soluble secretory extracellular domains, and effectively regulate the uptake of oxidized and acetylated low-density lipoprotein [146]. Scavenger receptor class A (SR-A) is expressed mainly on the surfaces of mature macrophages and has been associated with atherogenesis [147]. This receptor has been targeted for RA treatment: MTX-loaded layered double hydroxide nanocomposites have been modified with dextran sulfate (LDH-MTX-DS), a hydrophilic block that specifically binds SR-A [148]. LDH-MTX-DS releases MTX more rapidly at pH 5.5 than at pH 7.4, and actively targets scavenger receptors on the surfaces of activated RAW 264.7 cells, thus leading to significantly stronger therapeutic effects than those of free MTX in AIA rats [133]. In another study, micelles modified with dextran sulfate have also been prepared for celastrol delivery. These micelles have been found to effectively accumulate and release celastrol in RAW264.7 cells by binding SR-A, and to significantly improve the therapeutic effect of celastrol in vivo without causing clear systemic toxicity [134].
CD44 is a non-kinase transmembrane glycoprotein that is overexpressed in inflammatory synovial macrophages and fibroblasts [149]. It promotes pathological angiogenesis by regulating the proliferation, migration and adhesion of endothelial cells. HA is a natural polysaccharide in the extracellular matrix that specifically binds the CD44 receptor [54, 150]. Therefore, HA-coated acid-sensitive polymeric nanoparticles composed of egg phosphatidylcholine, polyethyleneimine and poly(cyclohexane-1,4-diyl acetone dimethylene ketal) have been loaded with dexamethasone to target activated macrophages overexpressing CD44 [135]. The obtained nanoparticles have been found to significantly decrease inflammatory cell infiltration as well as damage to bone and cartilage in the ankles of AIA rats, showing stronger therapeutic efficacy than the free drug [135]. In another study, HA-functionalized hydroxyapatite nanoparticles loaded with MTX and teriflunomide have shown high cellular uptake and cytotoxicity in vitro; prevented the progression of arthritis and promoted joint regeneration in CIA rats; and led to less hepatotoxicity than commercially available formulations [136]. Bilosomes loaded with tripterine, a natural compound with strong antioxidant, antiangiogenic and antirheumatic properties, have been prepared with cationic lipids through the thin film hydration method, then coated with HA to form HA@Tri-BLs [137]. The resulting nanosystem has been found to improve the circulation time of tripterine in vivo, and increase its systemic and intra-arthritic bioavailability, in a collagen antibody-induced arthritis model. The in vivo anti-arthritic efficacy of HA@Tri-BLs has also been found to be significantly higher than that of uncoated bilosomes, owing to high drug accumulation in the articular cavity.
Mannose receptors belong to the C-type lectin family and are overexpressed on the surfaces of macrophages during inflammation; they mediate endocytosis of glycoproteins [151]. Mannose-conjugated liposomes loaded with p-coumaric acid, a compound with anti-inflammatory and osteoclastic effects, prolongs the drug’s residence time in joints and downregulates pro-inflammatory TNF-α, IL-1β, IL-6 and IL-23, as well as the transcription factor NFATc1, which triggers osteoclast differentiation [138]. In another study, mannose-modified liposomes have been used to encapsulate morin, a bioflavonoid with anti-inflammatory, antitumor and anti-oxidant activity. These mannose-modified liposomes are preferentially internalized by macrophages from arthritic rats, and inhibit the inflammatory immune response and osteoclastogenesis better than the reference drug dexamethasone palmitate encapsulated in mannosylated liposomes, according to clinical and histological analysis [139].
5.3. Targeting based on environmental stimuli
The microenvironment of inflamed tissues differs from that of healthy tissue in pH, redox conditions, and the levels of oxygen, reactive oxygen species (ROS), glucose, enzymes and ATP. These differences can be exploited to trigger drug release from responsive delivery systems, thereby avoiding premature release into the circulation or healthy tissues, and decreasing the risk of adverse or other off-target effects [152–154].
5.3.1 pH-responsive drug delivery
Under normal conditions, the pH of the extracellular medium and blood is usually ∼7.4, but the pH falls to 6.0 in the synovial microenvironment [155]. This pH difference between the inflammatory microenvironment and normal tissues has been exploited to design pH-responsive targeted drug delivery systems based either on ionizable polymers (polyacids or polybases), whose conformation or solubility changes with pH, or on polymer systems with acid-sensitive bonds [155, 156]. For example, pH-sensitive polymeric micelles prepared through self-assembly of PEG-based derivatives and the hydrophobic drug prednisolone through acid-labile hydrazone bonds have been found to promote the accumulation of prednisolone in inflamed joints and to show stronger anti-inflammatory effects in vivo than the free drug [157]. Another study has reported the preparation of pH-sensitive polymer nanoparticles that deliver siRNA when the surrounding pH is 5.0, thus allowing selective drug delivery to sites of inflammation, and leading to therapeutic efficacy in AIA rats [52]. A small library of biocompatible amphiphilic polymers based on methoxy poly(ethylene glycol)-poly(cyclohexane-1,4-diyl acetone dimethyleneketal) and methoxy poly(ethylene glycol)-poly((cyclohexane86.7%, 1,5-pentane-diol13.3%)-1,4-diyl acetone dimethylene ketal) has been synthesized for the targeted delivery of superoxide dismutase. The novel polymers release the enzyme in a pH-dependent manner, thus allowing the enzyme, which is normally rapidly cleared or degraded in vivo, to show good anti-oxidant and anti-inflammatory activities in AIA rats [158].
5.3.2 Redox-responsive drug delivery
Activated T cells produce 10–100 times more ROS at sites of inflammation than in normal tissues. These ROS trigger an anti-oxidant glutathione (GSH) response, which prevents further increases in ROS and resultant cellular damage [159]. The increased GSH concentrations in inflammatory microenvironments result in cleavage of disulfide bonds [160, 161]. Reduction-responsive polyprodrug amphiphiles have been prepared through “reversible addition fragmentation chain transfer” polymerization of indomethacin-based redox-responsive prodrug monomers bearing disulfide bonds that are GSH responsive and phenylboronic acid ester bonds that are H2O2 responsive [162]. The resulting polymers have been found to efficiently antagonize the effects of lipopolysaccharide on RAW264.7 macrophages.
5.3.3 ROS-responsive drug delivery
ROS, such as H2O2, superoxide (O2 • −), hydroxyl radical (•OH), peroxynitrite (ONOO−) and hypochlorite (OCl−), play important roles in normal cellular signaling pathways and oxidative metabolism. However, their excessive production in cells or tissues can cause oxidative stress, thus leading to various diseases, including inflammation, cancer and atherosclerosis [154, 163, 164]. To target the high ROS levels in RA tissues, 4-phenylboronic acid pinacol ester-conjugated cyclodextrin biomaterials have been used to prepare ROS-responsive dexamethasone-loaded nanoparticles [165]. The particles have been found to be efficiently internalized by activated macrophages, to accumulate in inflamed joints, and to decrease joint swelling and cartilage destruction in CIA mice.
5.3.4 Enzyme-responsive drug delivery
Enzymes such as proteases, glycosidases, metalloproteases, lipases and phospholipases are biocatalysts that play key roles in countless normal processes [166], but their expression may be altered in disease [167]. Such changes can be exploited as triggers for environmentally responsive drug delivery [168]. For example, phospholipase A2 is overexpressed under inflammatory conditions, and it specifically hydrolyzes sn-2 ester bonds in phospholipids [169]. Thus, this enzyme attacks liposomes of the appropriate composition and consequently enables controlled release of drug cargo [170], Similarly, high phospholipase A2 levels can release colchicine from phosphatidylcholinase-responsive liposomes that are otherwise stable in the blood circulation [171].
5.4 Local injection strategies
Local intra-articular injection is also an important therapeutic strategy for RA. Intra-articular injection has several advantages in RA therapy, including good bioavailability, diminished systemic exposure and adverse events, and lower costs. The intra-articular injection of corticosteroids is the first-line treatment for RA. Researchers have found that intra-articular injection of etanercept is a safer and more promising treatment than corticosteroid treatment [172]. Drugs currently used for intra-articular injection are dissolved in solution. Consequently, the drug rapidly diffuses into the systemic circulation after intra-articular injection, thereby resulting in rapid removal from the joint cavity and short retention time. Frequent intra-articular injection is required for good therapeutic effects against RA, thus increasing the risk of local pain, joint swelling and infection [173]. Therefore, several nanoplatforms have been developed to improve drug accumulation at inflamed sites. For example, injection of tetramethylpyrazine as a nanosuspension with hydrophobic ions prolongs its retention in the articular cavity in rats and leads to greater anti-arthritic efficacy than that of the free drug [174]. Moreover siRNAs targeting TNF loaded into lipid-polymer hybrid nanoparticles of lipidoid and PLGA have been found to inhibit inflammation in murine experimental arthritis models, even at the low siRNA dose of 1 μg [175]. Intra-articular injection of HA-modified liposomes loaded with diclofenac and dexamethasone has been observed to effectively decrease inflammation and paw swelling in mice for 4 weeks [176].
6. SUMMARY AND PERSPECTIVES
Although great progress has been made in treating RA, the clinical application of traditional therapies is limited by high costs and requirements for frequent, long-term dosing. Nanocarriers can actively or passively deliver drugs to inflamed sites, thereby prolonging drug half-life, improving drug accumulation in target tissues and decreasing drug systemic toxicity. Although large numbers of nanocarriers have been developed for RA therapy, few have entered clinical trials. These nanocarriers face several challenges. (1) Most nanocarriers are unstable in the circulation and usually leak the drug before reaching inflamed sites. (2) Nanocarriers are easily captured by the reticuloendothelial system after intravenous injection. (3) The targeting activity of nanocarriers for inflamed regions is generally too low. Consequently, developing safer, more efficient nanocarriers remains the most important research frontier at present.
In addition, although nanocarriers can greatly improve therapeutic effects in RA, several shortcomings remain. For example, the modification of ligands or targeting molecules is complex and costly. How to simplify modification procedures and decrease costs will be key for clinical transformation. PEG modification is usually used to prolong the blood circulation time in RA targeting by nanocarriers. However, this modification can also hinder interactions between nanoparticles and cells, and inhibit cellular uptake. In addition, after multiple intravenous injections, the PEG-modified nanoparticles are rapidly removed from the blood circulation [177]. Therefore, developing new simply and easily prepared biological materials that can avoid phagocytosis by the reticuloendothelial system, prolong the circulation time and target arthritis sites will be critical for RA therapy.
Recent research has focused on the construction of targeting nanocarriers modified with ligands that bind specific receptors on the surfaces of cells involved in RA. Most of these nanocarriers target RA by targeting the receptors on vascular endothelial cells, fibroblast-like synoviocytes and macrophages. Beyond these cells, autoreactive T cells and B cells also substantially contribute to the inflammatory process. Activated T cells activate monocytes, macrophages and synovial fibroblasts. Developing nanocarriers targeting these cells will also be important.
A recent approach for targeted treatment of RA involves delivery of RA-associated antibodies and antigens to auto-reactive lymphocytes or dendritic cells, with the aim of inducing antigen-specific immune tolerance [178] while maintaining protective immunity [179, 180]. To this end, future work should explore as many RA-associated antibodies and antigens as possible. For example, RA-associated cell injury and death result in the release of cell-free DNA (cfDNA) into the peripheral blood and synovial fluid [181, 182]. Auto-antibodies bind the cfDNA, and the resulting complex activates TLRs, thereby leading to the secretion of inflammatory cytokines [183]. Thus, using cationic polymers to eliminate cfDNA may be a promising treatment against RA. In a step in this direction, one study has shown that cationic dimethylamino-group-modified polydopamine nanoparticles strongly bind cfDNA and efficiently inhibit cfDNA-induced inflammation in an animal model [184]. Decreasing the systemic toxicity of these cationic polymeric nanoparticles is an important priority for future work.
RA-associated disability and mortality currently affect large numbers of people. Despite decades of exploration of nanocarriers for RA treatment, many areas have yet to be investigated. Developing superior nanocarriers for RA therapy is important and urgently needed.