- Record: found
- Abstract: found
- Article: not found
Vaccinia Virus Proteins A52 and B14 Share a Bcl-2–Like Fold but Have Evolved to Inhibit NF-κB rather than Apoptosis
Read this article at

Abstract
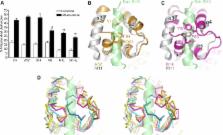
Vaccinia virus (VACV), the prototype poxvirus, encodes numerous proteins that modulate the host response to infection. Two such proteins, B14 and A52, act inside infected cells to inhibit activation of NF-κB, thereby blocking the production of pro-inflammatory cytokines. We have solved the crystal structures of A52 and B14 at 1.9 Å and 2.7 Å resolution, respectively. Strikingly, both these proteins adopt a Bcl-2–like fold despite sharing no significant sequence similarity with other viral or cellular Bcl-2–like proteins. Unlike cellular and viral Bcl-2–like proteins described previously, A52 and B14 lack a surface groove for binding BH3 peptides from pro-apoptotic Bcl-2–like proteins and they do not modulate apoptosis. Structure-based phylogenetic analysis of 32 cellular and viral Bcl-2–like protein structures reveals that A52 and B14 are more closely related to each other and to VACV N1 and myxoma virus M11 than they are to other viral or cellular Bcl-2–like proteins. This suggests that a progenitor poxvirus acquired a gene encoding a Bcl-2–like protein and, over the course of evolution, gene duplication events have allowed the virus to exploit this Bcl-2 scaffold for interfering with distinct host signalling pathways.
Author Summary
Cells possess formidable defences against virus infection, but viruses have evolved sophisticated counter-measures to evade such defences. Vaccinia virus, the vaccine used to eradicate smallpox, has about 200 genes, and many of these encode proteins that help the virus evade the host's immune defences. This paper concerns the vaccinia virus proteins A52 and B14, which block signalling pathways leading to the activation of the NF-κB transcription factor and thereby diminish the host immune response to infection. By solving the three-dimensional structures of A52 and B14, we show that they closely resemble a family of cellular and viral proteins (the Bcl-2 family) that usually function to regulate apoptosis (a process by which cells commit suicide, thereby stopping the replication of any viruses with which they are infected). However, neither A52 nor B14 regulate apoptosis. By comparing three-dimensional structures, we show that vaccinia virus Bcl-2–like proteins more closely resemble each other than they do other cellular or viral Bcl-2–like proteins. This suggests that an ancestor of vaccinia virus acquired a gene encoding a Bcl-2–like protein from its host and, over time, this gene has been copied and adapted for different functions within the virus.
Related collections
Most cited references52
- Record: found
- Abstract: found
- Article: not found
Automated structure solution with autoSHARP.
- Record: found
- Abstract: found
- Article: not found
Substructure solution with SHELXD.
- Record: found
- Abstract: found
- Article: not found