- Record: found
- Abstract: found
- Article: found
Fibroblast-induced switching to the mesenchymal-like phenotype and PI3K/mTOR signaling protects melanoma cells from BRAF inhibitors
Read this article at
Abstract
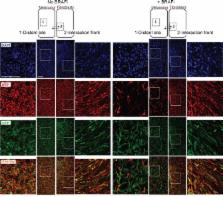
The knowledge on how tumor-associated stroma influences efficacy of anti-cancer therapy just started to emerge. Here we show that lung fibroblasts reduce melanoma sensitivity to the BRAF inhibitor (BRAFi) vemurafenib only if the two cell types are in close proximity. In the presence of fibroblasts, the adjacent melanoma cells acquire de-differentiated mesenchymal-like phenotype. Upon treatment with BRAFi, such melanoma cells maintain high levels of phospho ribosomal protein S6 (pS6), i.e. active mTOR signaling, which is suppressed in the BRAFi sensitive cells without stromal contacts. Inhibitors of PI3K/mTOR in combination with BRAFi eradicate pS6 high cell subpopulations and potentiate anti-cancer effects in melanoma protected by the fibroblasts. mTOR and BRAF co-inhibition also delayed the development of early-stage lung metastases in vivo. In conclusion, we demonstrate that upon influence from fibroblasts, melanoma cells undergo a phenotype switch to the mesenchymal state, which can support PI3K/mTOR signaling. The lost sensitivity to BRAFi in such cells can be overcome by co-targeting PI3K/mTOR. This knowledge could be explored for designing BRAFi combination therapies aiming to eliminate both stroma-protected and non-protected counterparts of metastases.
Related collections
Most cited references27
- Record: found
- Abstract: found
- Article: not found
Final version of 2009 AJCC melanoma staging and classification.
- Record: found
- Abstract: found
- Article: not found
Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation.
- Record: found
- Abstract: found
- Article: not found