- Record: found
- Abstract: found
- Article: found
Vitamin E δ-tocotrienol triggers endoplasmic reticulum stress-mediated apoptosis in human melanoma cells
Read this article at
Abstract
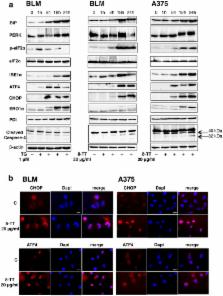
Malignant melanoma is the leading cause of death from skin cancer. Drug toxicity and resistance represent a serious challange for melanoma treatments. Evidence demonstrates that natural compounds may play a crucial role in cancer prevention, growth and progression. Vitamin E tocotrienols (TT) were shown to possess antitumor activity. Here, we analyzed the effects of δ-TT on melanoma cell growth and the involvement of the endoplasmic reticulum (ER) stress in this activity. The experiments were performed on human melanoma cell lines, BLM and A375. δ-TT exerted a significant proapoptotic effect on both cell lines, involving the intrinsic apoptosis pathway; importantly, this compound did not affect the viability of normal human melanocytes. In melanoma cells, δ-TT exerted its antitumor effect through activation of the PERK/p-eIF2α/ATF4/CHOP, IRE1α and caspase-4 ER stress-related branches. Salubrinal, an inhibitor of the ER stress, counteracted the cytotoxic activity of δ-TT. In vivo experiments performed in nude mice bearing A375 xenografts evidenced that δ-TT reduces tumor volume and tumor mass; importantly, tumor progression was significantly delayed by δ-TT treatment. In conclusion, δ-TT exerts a proapoptotic activity on melanoma cells, through activation of the ER stress-related pathways. δ-TT might represent an effective option for novel chemopreventive/therapeutic strategies for melanoma.
Related collections
Most cited references47
- Record: found
- Abstract: found
- Article: not found
ER stress-induced cell death mechanisms.
- Record: found
- Abstract: not found
- Article: not found
Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma.
- Record: found
- Abstract: found
- Article: not found