- Record: found
- Abstract: found
- Article: found
An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E -/- mice
Read this article at
Abstract
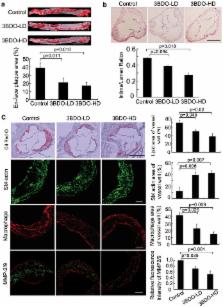
Oxidized low-density lipoprotein (oxLDL) inhibits mammalian target of rapamycin (mTOR) and induces autophagy and apoptosis in vascular endothelial cells (VECs) that play very critical roles for the cardiovascular homostasis. We recently defined 3-benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran-2(3H)-one (3BDO) as a new activator of mTOR. Therefore, we hypothesized that 3BDO had a protective role in VECs and thus stabilized atherosclerotic lesions in apolipoprotein E -/- (apoE -/-) mice. Our results showed that oxLDL inhibited the activity of mTOR and increased the protein level of autophagy-related 13 (ATG13) and its dephosphorylation, thus inducing autophagy in human umbilical vein endothelial cells (HUVECs). All of these effects were strongly inhibited by 3BDO. In vivo experiments confirmed that 3BDO activated mTOR and decreased the protein level of ATG13 in the plaque endothelium of apoE -/- mice. Importantly, 3BDO did not affect the activity of mTOR and autophagy in macrophage cell line RAW246.7 and vascular smooth muscle cells of apoE -/- mice, but suppressed plaque endothelial cell death and restricted atherosclerosis development in the mice. 3BDO protected VECs by activating mTOR and thus stabilized atherosclerotic lesions in apoE -/- mice.
Related collections
Most cited references33
- Record: found
- Abstract: found
- Article: not found
Autophagy in cell death: an innocent convict?
- Record: found
- Abstract: found
- Article: not found
A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy.
- Record: found
- Abstract: found
- Article: not found